Active muscles store vitamin D in the winter
The Role of Skeletal Muscle in Maintaining Vitamin D Status in Winter
Curr Dev Nutr. 2019 Jul 25;3(10):nzz087. doi: 10.1093/cdn/nzz087. eCollection 2019 Oct.
Rebecca S Mason ,1 Mark S Rybchyn©1 Myriam Abboud©1'3 Tara C Brennan-Speranza ,1 and David R Fraser©2
1 Department of Physiology, School of Medical Sciences and Bosch Institute;
2 Sydney School of Veterinary Science, Faculty of Science, The University of Sydney, NSW 2006, Australia; and
3 Zayed University, Dubai, United Arab Emirates
📄 Download the PDF from VitaminDWiki
The status of vitamin D is determined mainly by its formation in skin by the photochemical action of solar UVB light (wavelength 290-320 nm) on the precursor 7-dehydrocholesterol. Because of seasonal variation in intensity of solar UV light, vitamin D status falls in winter and rises in summer. It has been presumed that there is no functional store of vitamin D. Thus, to avoid deficiency, a nutritional supply would be required in winter. However, there is now evidence that the main circulating metabolite of vitamin D, 25-hydroxyvitamin D, accumulates in skeletal muscle cells, which provide a functional store during the winter months. The mechanism is mediated by muscle cell uptake of circulating vitamin D-binding protein (DBP) through a megalin-cubilin membrane transport process. DBP then binds to cytoplasmic actin to provide an array of high-affinity binding sites for 25-hydroxyvitamin D [25(OH)D]. The repeated passage of 25(OH)D into and out of muscle cells would account for its long residence time in blood.
In 1918, Sir Edward Mellanby (1) published his conclusion that there was a nutritional factor in meat extracts, cod liver oil, and butter that prevented the development of experimental rickets in beagle pups fed on a bizarre diet for carnivorous animals, of rice, oatmeal, and milk. That study was undertaken during the years when micronutrients in general were being discovered. It seemed therefore that this antirachitic factor, subsequently named vitamin D, was one of several essential micronutrients to be obtained from food. But, if a 1921 report by Hess and Unger (2) of sunlight exposure curing infantile rickets, had preceded Mellanby’s brief publication, perhaps the concept that vitamin D was a nutrient might not have become so firmly established.
Later research established that vitamin D is produced in skin by the photochemical action of solar UVB radiation (wavelengths 290-320 nm) on its precursor, 7-dehydrocholesterol (3). After transport in blood to the liver, vitamin D is converted to 25-hydroxyvitamin D [25(OH)D] and then in the kidney, in a functionally regulated manner, to the hormone, 1,25-dihydroxyvitamin D [1,25(OH)2D]. This acts as a steroid hormone in calcium homeostasis and in many other physiological processes (4).
In the 1970s, a method of estimating the vitamin D status of individuals became available. It was found that the concentration of the metabolite, 25(OH)D, in blood serum or plasma, was positively related to the supply of vitamin D, either from exposure of skin to sunlight or to the quantity consumed by mouth (5). Although vitamin D is widely considered to be a nutrient, natural foo ds contain only minute amounts of it, with fish, not just fatty fish (5), and eggs from hens fed vitamin D-fortified diets, having nutritionally significant quantities. It became apparent that for most terrestrial vertebrates, vitamin D is obtained by exposure of the skin to solar UVB light. Measurement of 25(OH)D concentration in blood revealed a seasonal variation, with the highest concentrations being found in summer and the lowest in winter (7). This seasonal variation in vitamin D status correlated well with the seasonal variation in the intensity of solar UVB light (8, 9). Despite the concept that vitamin D was a nutrient, it became clear that the vitamin D status of populations was largely determined by exposure to solar UVB light rather than by dietary intake of vitamin D in food (10, 11). Furthermore, it became apparent that in winter the vitamin D status of many people was suboptimal or deficient (12).
For many vertebrates living in temperate regions of the world, there is no solar UVB radiation on their skin for part or all of winter. Therefore, for humans, the concept has developed that a dietary source of vitamin D is needed in winter, to avoid deficiency when no vitamin D is being produced in skin (12). Yet nonhuman vertebrates, in the same environment, do not obtain supplementary dietary sources of vitamin D and in general do not become functionally deficient. Could there be a storage mechanism that allows vitamin D function to be maintained in winter and if so, why do so many humans become deficient in winter? Compared with the true fat-soluble micronutrients, vitamin D does not seem to have a definable tissue storage site (13). Unlike, for example, vitamin A, which is convincingly stored in the liver, the vitamin D content of that organ is very low and represents only that which is in the process of being metabolized to 25(OH)D or to breakdown products to be excreted in bile (14). In the search for a storage site, there have been many reports of vitamin D accumulating in adipose tissue, and this has therefore been assumed to be the tissue of storage (see, e.g., references 15, 16). Although vitamin D appears to be released when weight is lost and adipose tissue volume decreases (17), vitamin D is highly lipophilic and, to our knowledge, no specific mechanism has been found that would make adipose tissue a functional store so that vitamin D, trapped in the cytoplasmic lipid of adipocytes, could be mobilized when a deficiency of vitamin D was developing. The highest tissue concentration of any molecular form of vitamin D is that of 25(OH)D in blood. Thus, the blood circulation has also been assumed to be the storage site of vitamin D, particularly because the residence time half-life of 25(OH)D ranges from 15 to 60 d (18) and perhaps even as long as 120 d (19). This is far longer than a typical steroid in blood such as estradiol, with a half-life of only 2-3 d (20), or indeed of the endocrine product of vitamin D, 1,25(OH)2D, with a half-life of only 5-8 h (21, 22)
However, there is a problem with this strange concept that blood acts as a storage site for vitamin D as its metabolite, 25(OH)D. In the circulation, 25(OH)D is transported, bound with high affinity to a specific vitamin D-binding protein (DBP) with a single binding site per molecule of protein. The same protein is also the vehicle in blood for transporting parent vitamin D and the hormonal product, 1,25(OH)2D 23). Whereas 25(OH)D has a residence half-life of many days, the half-life of DBP is much shorter at 1-4 d (24). Furthermore, with the normal concentration of 25(OH)D of 50-100 nmol/L, only 1-3% of DBP molecules, at a concentration of 5-7 pmol/L (25), would have 25(OH)D attached to the specific binding site. Therefore, each 25(OH)D molecule, to maintain its long half-life in blood, would have to transfer many times from one DBP to another. No mechanism has yet been found to explain how such a process could operate.
Alternatively, to account for its long residence half-life, each molecule of 25(OH)D could repeatedly passage to and from some extravascular site, before ultimately either being converted to the active hormone, 1,25(OH)2D, or else being metabolically destroyed in the liver. Vitamin D metabolites clearly have a functional role in muscle, though this is poorly understood at a molecular level (26). As part of this functional role or in addition to this role, various studies in vivo have suggested that skeletal muscle might be the extravascular tissue into which 25(OH)D in blood passes, and then after some time returns to the circulation.
When radioactively labeled vitamin D was administered to pregnant rats, most of the radioactivity recovered in the newborn pups was found in skeletal muscle as 25(OH)D (27) . This could be a reserve to meet requirements in early neonatal life, when vitamin D supply from the environment would be minimal. It is also possible that regular physical exercise might have an influence on maintaining an adequate concentration of 25(OH)D in blood. A cross-sectional survey of 323 adolescent girls, living at a latitude of 40o N, found that the concentration of 25(OH)D in blood plasma at the end of winter was significantly higher in those undertaking regular physical exercise compared with those who led a more sedentary lifestyle (28). This finding might suggest that the extent of physical exercise was simply an indicator of the time spent outdoors exposed to solar UVB light, which could account for the positive association between exercise and vitamin D status. However, physical exercise indoors was also reported to be associated with higher 25(OH)D serum levels (29), and this was shown to be independent of the amount of sun exposure (30) .
These results (27-30) suggested that muscle cells have the ability to take up 25(OH)D from blood and that the capacity to do so is increased by some change associated with regular physical exercise . DBP in the circulation is synthesized and secreted by the liver. This protein has 2 specific, high-affinity binding sites. One is for vitamin D and its metabolites, with the highest affinity being for 25(OH)D (Kd < 1 nM). One of the curious features of DBP is that it has an additional binding site for actin (31). Various investigators have proposed that this functions as a scavenger of actin, which might be released into the circulation when there is cellular injury (32, 33). A commonly held theory postulates that the actin-binding site of DBP functions to bind actin if the latter is released into blood from damaged cells, and thus protects against intravascular coagulation. Yet it was known over 30 y ago that DBP becomes tightly bound to actin in skeletal muscle (34). This raised the question as to whether DBP might be incorporated into muscle cells via a cell membrane megalin-mediated process, in a similar fashion to the incorporation of DBP into hepatic stellate cells and its subsequent binding to intracellular actin (35). We proposed that the plasma membrane ofskeletal muscle cells contains the proteins megalin and cubulin and that these function to transfer extracellular DBP into the cytoplasm of muscle cells where it binds to actin filaments. In this way skeletal muscle cells can contain an array of high-affinity binding sites for 25(OH)D in the form of DBP bound to actin. Small amounts of unbound 25(OH)D in the extracellular fluid can, like other hormonal steroids, diffuse through the lipid bilayer of the plasma membrane, into and out of muscle cells. Intracellular DBP, bound to actin, thus would provide a mechanism for the accumulation and retention of 25(OH)D in skeletal muscle. However, it has been shown in vivo in rabbits that DBP in muscle has a short residence time and undergoes proteolytic degradation. When DBP is thus destroyed, any bound 25(OH)D would be released and could then diffuse from the cell and eventually back into the circulation. This process of 25(OH)D passaging into and out of muscle cells therefore would explain the long residence time for this metabolite in the circulation (Figure 1). It is possible to speculate that the muscle storage of 25(OH)D evolved secondary to a function for 25(OH)D in muscle, because an actin-binding site on DBP is not present in fish, and perhaps not in amphibians, but is present in reptiles, birds, and mammals (36).
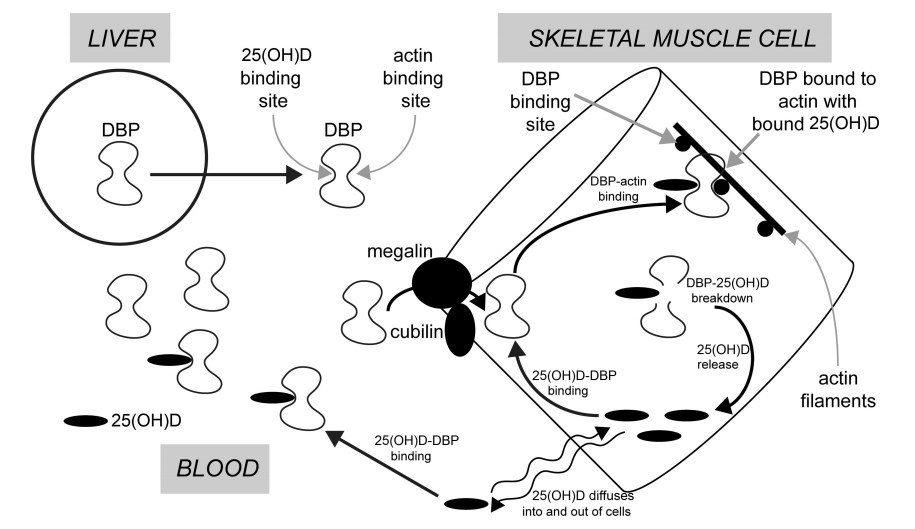
- FIGURE 1 The mechanism by which vitamin D–binding protein (DBP) from blood could be internalized into skeletal muscle cells to provide high-affinity intracellular binding sites for 25-hydroxyvitamin D [25(OH)D]. It is postulated that this intracellular DBP enables 25(OH)D, which diffuses into muscle cells, to be bound and retained until the DBP undergoes proteolysis. The released 25(OH)D then diffuses from the cell and is immediately bound by extracellular DBP and then returns to the circulation
This process has been investigated with cells in culture (37). When the uptake and retention of labeled 25(OH)D was measured in 1) undifferentiated C2C12 murine myoblasts, 2) these cells after differentiation into myotubes, and 3) MG63 osteoblasts as a nonmuscle cell control, the myotubes were the only cell type to accumulate 25(OH)D (Figure 2). A similar result was obtained with isolated primary mouse muscle cells (37). The specific affinity for 25(OH)D by the differentiated muscle cells was also revealed by their ability to retain labeled 25(OH)D when placed in a medium devoid of 25(OH)D. In contrast, the control osteoblasts and undifferentiated myoblasts, under these conditions, rapidly released into the medium most of the small amount of previously acquired 25(OH)D.
The mechanism of uptake and accumulation of 25(OH)D by muscle cells became apparent when immunohistochemistry revealed megalin, and its associated protein cubulin, in the cell membrane, and DBP was visualized in the cell cytoplasm (37). Furthermore, when Alexafluor- 488-labeled DBP (Molecular Probes, Oregon, USA; Merck, Darmstadt, Germany) was added to the medium, differentiated muscle cells took up this protein by endocytosis, and confocal microscopy showed it to be in close association with the cytoplasmic actin filaments. The specific role of megalin in transmembrane uptake of extracellular DBP to provide intracellular binding sites for 25(OH)D was also demonstrated when myotubes, treated with receptor-associated protein, an inhibitor of megalin function, showed diminished ability to accumulate labeled 25(OH)D (37). From these results it is concluded that mature muscle cells have a specific mechanism for taking up DBP, which when bound to actin acts as an intracellular retention site for 25(OH)D that diffuses into the cells from the extracellular fluid.
For muscle cells to act as either a storage site or an extravascular recycling site for 25(OH)D, there would need to be some regulating factor or factors that would enhance either its uptake or release from these cells. One candidate for such a regulator could be parathyroid hormone (PTH), because the serum concentration of this hormone increases slightly when the serum concentration of 25(OH)D declines below 50-60 nmol/L (28). The PTH receptor was identified in the cell membrane of myotubes (Figure 3A), and low concentrations of PTH (0.1-10 pM) in the medium of myotubes that had accumulated labeled 25(OH)D provoked release of 25(OH)D. After 4 h in vehicle, approximately 74% of tritium was retained in the cells, and 61% after 8 h. In contrast, retention of tritiated 25(OH)D3 by myotubes, in the presence of PTH, decreased with time in a concentration-dependent manner. (Figure 3B) (4). Paradoxically, 1,25(OH)2D also modified both 25(OH)D uptake and its release from both myotubes and primary myotubes in vitro (38). It is suggested that proteolysis of the intracellular DBP allows the released 25(OH)D to diffuse out of the cell, where it would be readily bound again by DBP in the extracellular fluid.
Identification of the physiological mechanism that allows uptake and release of 25(OH)D by skeletal muscle cells awaits further studies in vivo. But there is evidence in sheep that muscle accumulates 25(OH)D in winter as vitamin D status is falling, and that this higher concentration of 25(OH)D declines to the concentrations of summer, when the circulating concentration of 25(OH)D rises. In preliminary studies, 2 sheep grazing on pasture throughout the year had 25(OH)D concentrations in blood in the range 25-30 nmol/L at the end of winter, compared with the typical concentration in sheep in summer of about 50 nmol/L (4). However, in contrast to the low concentration of 0.1-0.2 fig 25(OH)D/100 g wet weight of muscle in summer, analysis of biopsies of skeletal muscle of these sheep in winter revealed that the concentration of 25(OH)D was 30 to 70 times greater at 6.8-7 ug/100 g. When these sheep were each given an oral dose of 1.25 mg 25(OH)D (at day 0), the plasma concentration predictably rose to values, 5 d later, of ~150 nmol/L. Surprisingly, however, this elevation of vitamin D status resulted in a gradual fall in the concentration of 25(OH)D in muscle biopsies, so that after 40 d the 25(OH)D concentrations in both muscle and plasma had declined to the usual levels found in summer. It seems, therefore, that when vitamin D status improves, the ability of muscle cells to accumulate large quantities of 25(OH)D is lost (Figure 4) (4, 39).
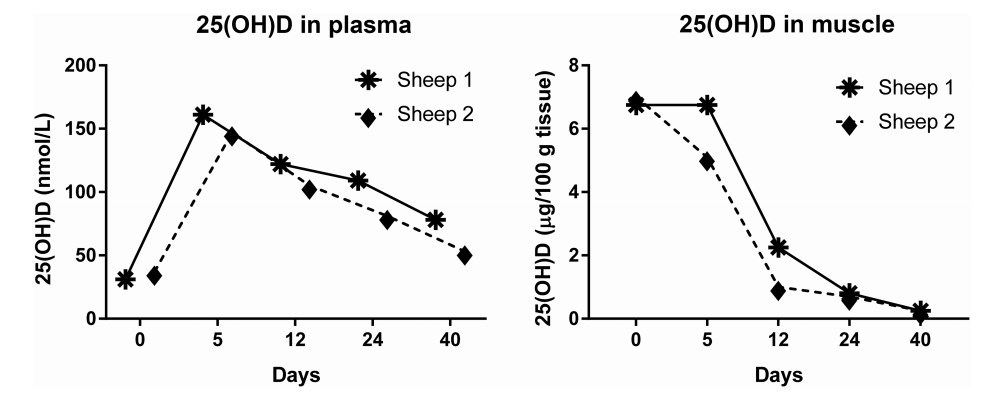
- FIGURE 4 High concentrations of 25-hydroxyvitamin D [25(OH)D] in skeletal muscle of sheep at the end of winter decline as vitamin D
status rises. Plasma concentrations of 25(OH)D in 2 sheep on pasture at the end of winter and after an oral dose of 1.25 mg 25(OH)D
(at day 0). Concentrations of 25(OH)D in muscle biopsies taken at the same time. Figure redrawn from reference 4
If skeletal muscle has the function of conserving 25(OH)D and regulating its concentration in blood, why then is vitamin D deficiency so frequently found in people during the winter months, whereas animals in the same environment have apparently adequate vitamin D status? One obvious reason would be that many humans have an indoor lifestyle and do not build up adequate levels of vitamin D during summer, by exposure of their skin to solar UVB radiation. However, compared with nondomestic animals, many humans also lead a rather sedentary life. Epidemiological studies suggest that regular physical exercise is positively associated with the concentrations of 25(OH)D in blood (28-30), thus indicating that muscle energy metabolism could be important for its role in maintaining adequate vitamin D status. Epidemics of vitamin D-deficiency rickets have been reported in young children who are afflicted with protein and energy malnutrition because of restricted food supply (39) or from poverty (40). Because of the severe effect of protein/energy malnutrition on skeletal muscle function (41), this again suggests that defective muscle energy metabolism leads to a defect in the role of skeletal muscle in conserving 25(OH)D when vitamin D supply from the environment ceases in winter.
General Conclusion
The research summarized here explores the hypothesis that 25(OH)D has an apparently long residence time in the circulation because it passages into and out of muscle cells under the influence of regulated internalization of DBP. There is evidence that this process is enhanced in winter, which could enable adequate vitamin D status to be maintained during the seasonal cessation of vitamin D supply from solar irradiation of skin. It is proposed that such a mechanism can become defective when muscle function declines with lack of exercise or malnutrition . Future research could explore in vivo the endocrine mechanism regulating the uptake and release of 25(OH)D from muscle cells as well as the mechanism by which disturbances of muscle energy metabolism appear to be associated with an inability to maintain adequate vitamin D status in winter.
References
Mellanby E. The part played by an “accessory factor” in the production of experimental rickets. J Physiol 1918;52:xi-xii.
Hess AF, Unger LJ. The cure of infantile rickets by sunlight. J Am Med Assoc 1921;77:39.
Havinga E. Vitamin D, example and challenge. Experientia 1973;29:1181 — 93.
Abboud M, Rybchyn MS, Liu J, Ning Y, Gordon-Thomson C, Brennan-Speranza TC, Cole L, Greenfield H, Fraser DR, Mason RS. The effect of parathyroid hormone on the uptake and retention of 25-hydroxyvitamin D in skeletal muscle cells. J Steroid Biochem Mol Biol 2017;173:173-9.
Tucker G 3rd, Gagnon RE, Haussler MR. Vitamin D 3 -25-hydroxylase: tissue occurrence and apparent lack of regulation. Arch Biochem Biophys 1973;155:47-57.
Padula D, Greenfield H, Cunningham J, Kiermeier A, McLeod C. Australian seafood compositional profiles: a pilot study. Vitamin D and mercury content. Food Chem 2016;193:106-11.
King L, Dear K, Harrison SL, van der Mei I, Brodie AM, Kimlin MG, Lucas RM. Investigating the patterns and determinants of seasonal variation in vitamin D status in Australian adults: the Seasonal D Cohort Study. BMC Public Health 2016;16:892.
O’Neill CM, Kazantzidis A, Ryan MJ, Barber N, Sempos CT, Durazo-Arvizu RA, Jorde R, Grimnes G, Eiriksdottir G, Gudnason V, et al. Seasonal changes in vitamin D-effective UVB availability in Europe and associations with population serum 25-hydroxyvitamin D. Nutrients 2016;8:E533.
Yu HJ, Kwon MJ, Woo HY, Park H. Analysis of 25-hydroxyvitamin D status according to age, gender, and seasonal variation. J Clin Lab Anal 2016;30: 905-11.
Ashwell M, Stone EM, Stolte H, Cashman KD, Macdonald H, Lanham-New S, Hiom S, Webb A, Fraser D. UK Food Standards Agency Workshop Report: an investigation of the relative contributions of diet and sunlight to vitamin D status. Br J Nutr 2010;104:603-11.
Lips P, van Schoor NM, de Jongh RT. Diet, sun, and lifestyle as determinants of vitamin D status. Ann N Y Acad Sci 2014;1317:92-8.
Mendes MM, Darling AL, Hart KH, Morse S, Murphy RJ, Lanham-New SA. Impact of high latitude, urban living and ethnicity on 25-hydroxyvitamin D status: a need for multidisciplinary action? J Steroid Biochem Mol Biol 2019;188:95-102.
Mawer EB, Backhouse J, Holman CA, Lumb GA, Stanbury SW. The distribution and storage of vitamin D and its metabolites in human tissues. Clin Sci 1972;43:413-31.
Bell PA, Kodicek E. Investigations on metabolites of vitamin D in rat bile. Separation and partial identification of a major metabolite. Biochem J 1969;115:663-9.
Lawson DE, Douglas J, Lean M, Sedrani S. Estimation of vitamin D3 and 25-hydroxyvitamin D3 in muscle and adipose tissue of rats and man. Clin Chim Acta 1986;157:175-81.
Brouwer DA, van Beek J, Ferwerda H, Brugman AM, van der Klis FR, van der Heiden HJ, Muskiet FA. Rat adipose tissue rapidly accumulates and slowly releases an orally-administered high vitamin D dose. Br J Nutr 1998;79:527-32.
Connors MH, Sheikholislam BM, Irias JJ. Vitamin D toxicity after dieting in hypoparathyroidism. Pediatrics 1976;57:794-6.
Clements MR, Davies M, Fraser DR, Lumb GA, Mawer EB, Adams PH. Metabolic inactivation of vitamin-D is enhanced in primary hyperparathyroidism. Clin Sci 1987;73:659-64.
Datta P, Philipsen PA, Olsen P, Bogh MK, Johansen P, Schmedes AV, Morling N, Wulf HC. The half-life of 25(OH)D after UVB exposure depends on gender and vitamin D receptor polymorphism but mainly on the start level. Photochem Photobiol Sci 2017;16:985-95.
Ginsburg ES, Gao X, Shea BF, Barbieri RL. Half-life of estradiol in postmenopausal women. Gynecol Obstet Invest 1998;45:45-8.
Gray RW, Caldas AE, Wilz DR, Lemann J Jr, Smith GA, DeLuca HF. Metabolism and excretion of 3H-1,25-(OH)2-vitamin D3 in healthy adults. J Clin Endocrinol Metab 1978;46:756-65.
Mason RS, Lissner D, Posen S, Norman AW. Blood concentrations of dihydroxylated vitamin D metabolites after an oral dose. Br Med J 1980;280:449-50.
Van Baelen H, Allewaert K, Bouillon R. New aspects of the plasma carrier protein for 25-hydroxycholecalciferol in vertebrates. Ann N Y Acad Sci 1988;538:60-8.
Haddad JG, Fraser DR, Lawson DEM. Vitamin-D plasma-binding protein—turnover and fate in the rabbit. J Clin Invest 1981;67:1550-60.
Bouillon R, Van Assche FA, Van Baelen H, Heyns W, De Moor P. Influence of the vitamin D-binding protein on the serum concentration of 1,25-dihydroxyvitamin D3. Significance of the free 1,25-dihydroxyvitamin D3 concentration. J Clin Invest 1981;67:589-96.
Gunton JE, Girgis CM. Vitamin D and muscle . Bone Rep 2018;8:163-7.
Clements MR, Fraser DR. Vitamin D supply to the rat fetus and neonate. J Clin Invest 1988;81:1768-73.
Foo LH, Zhang Q, Zhu K, Ma G, Trube A, Greenfield H, Fraser DR. Relationship between vitamin D status, body composition and physical exercise of adolescent girls in Beijing. Osteoporos Int 2009;20:417-25.
Bell NH, Godsen RN, Henry DP, Shary J, Epstein S. The effects of muscle -building exercise on vitamin D and mineral metabolism. J Bone Miner Res 1988;3:369-73.
Scragg R, Holdaway I, Jackson R, Lim T. Plasma 25-hydroxyvitamin D3 and its relation to physical activity and other heart disease risk factors in the general population. Ann Epidemiol 1992;2:697-703.
Van Baelen H, Bouillon R, De Moor P. Vitamin D-binding protein (Gc-globulin) binds actin. J Biol Chem 1980;255:2270-2.
Goldschmidt-Clermont PJ, Van Baelen H, Bouillon R, Shook TE, Williams MH, Nel AE, Galbraith RM. Role ofgroup-specific component (vitamin D binding protein) in clearance of actin from the circulation in the rabbit. J Clin Invest 1988;81:1519-27.
Otterbein LR, Cosio C, Graceffa P, Dominguez R. Crystal structures of the vitamin D-binding protein and its complex with actin: structural basis of the actin-scavenger system. Proc Natl Acad Sci U S A 2002;99: 8003-8.
Haddad JG. Human serum binding protein for vitamin D and its metabolites (DBP): evidence that actin is the DBP binding component in human skeletal muscle . Arch Biochem Biophys 1982;213:538-44.
Gressner OA, Lahme B, Gressner AM. Gc-globulin (vitamin D binding protein) is synthesized and secreted by hepatocytes and internalized by hepatic stellate cells through Ca(2+)-dependent interaction with the megalin/gp330 receptor. Clin Chim Acta 2008;390:28-37.
Bouillon R, Suda T. Vitamin D: calcium and bone homeostasis during evolution. Bonekey Rep 2014;3:480.
Abboud M, Puglisi DA, Davies BN, Rybchyn M, Whitehead NP, Brock KE, Cole L, Gordon-Thomson C, Fraser DR, Mason RS. Evidence for a specific uptake and retention mechanism for 25-hydroxyvitamin D (25OHD) in skeletal muscle cells. Endocrinology 2013;154:3022-30.
Abboud M, Rybchyn MS, Ning YJ, Brennan-Speranza TC, Girgis CM, Gunton JE, Fraser DR, Mason RS. 1,25-Dihydroxycholecalciferol (calcitriol) modifies uptake and release of25-hydroxycholecalciferol in skeletal muscle cells in culture. J Steroid Biochem Mol Biol 2018;177:109-15.
Chick DH. Study of rickets in Vienna 1919-1922. Med Hist 1976;20:41-51.
Fraser DR. Vitamin D-deficiency in Asia. J Steroid Biochem Mol Biol 2004;89-90:491-5.
Whitehead RG, Alleyne GA. Pathophysiological factors of importance in protein-calorie malnutrition. Br Med Bull 1972;28:72-9.
{include}