Vitamin D, immunity and microbiome
Vitamin D and immunity
Special section of interest: Why doesn’t vitamin D supplementation decrease disease risks?
F1000Prime Rep2014, 6:118 (doi: 10.12703/P6-118)),
Published: 01 Dec 2014 © 2014 Faculty of 1000 Ltd
Robyn M. Lucas1,2 robyn.lucas@telethonkids.org.au, Shelley Gorman1, Sian Geldenhuys1 and Prue H. Hart1
Telethon Kids Institute, University of Western Australia, 100 Roberts Road, Subiaco, Perth, Australia 6008
National Centre for Epidemiology and Population Health, The Australian National University, Canberra, Australia 0200
Abstract
Vitamin D deficiency has been linked to an increased risk of a wide range of adverse health outcomes. The active form of vitamin D has an important role in calcium metabolism and in bone mineralisation, but the evidence for other health outcomes is mixed, with the strongest effects seen in the weakest epidemiological study designs. There are plausible pathways whereby vitamin D deficiency can impair immune function, resulting in both overactivity and increased risk of autoimmune disease, as well as immune suppression with poorer resistance to infection. Vitamin D status may influence the bacterial flora that constitute the microbiome and affect immune function through this route. Exposure of the skin to ultraviolet radiation causes the production of a range of chemicals, including vitamin D, and new research is exploring possible vitamin D-independent immunomodulatory pathways.
📄 Download the PDF from VitaminDWiki.
Introduction
Vitamin D deficiency is reportedly widespread across the world, a consequence of urbanisation and indoor lifestyles, migration of dark-skinned populations to low sun environments when their skin type has evolved to be optimal for high sun environments, and possibly sun protection strategies to curb rising skin cancer incidence rates. Over the last 10-15 years, increased risk of a wide range of health outcomes has been linked to vitamin D deficiency, although in many cases the links remain rather weak. After the undisputed importance of vitamin D for bone health, the strongest evidence is probably for an increased risk of immune disorders in association with vitamin D deficiency. Here we briefly review some of that evidence, examine recent literature on possible mechanistic pathways and propose potential explanations for some of the conflicting results in this area.
Clues from epidemiology
Indications that vitamin D may be associated with disorders of human immune function often originate from observations of geographic variation in disease occurrence. In many regions of the world, vitamin D is primarily synthesised in the skin following sun exposure (specifically UV-B irradiation). Levels of UV-B radiation vary strongly according to distance from the equator (latitude) and time of year, with higher levels as the sun is closer to being directly overhead, that is, nearer the equator, in summer, and during the middle of the day. Thus, higher latitude is often taken as a proxy for both lower levels of ultraviolet radiation (UVR) and lower vitamin D status. Latitudinal gradients, where the incidence or prevalence of a disease increases with increasing distance from the equator (lower UV-B radiation), are described for multiple sclerosis [1], type 1 diabetes [2], the autoimmune vasculitides [3], the inflammatory bowel diseases [4], and asthma [5]. Null or inverse associations are also described for other autoimmune disorders [6,7]. Both UVR and vitamin D (in its active form 1,25(OH)2D, see Figure 1) have immunomodulatory effects [8] that provide a plausible mechanism whereby higher levels could decrease the risk of autoimmune diseases (see section on mechanisms, below) and, through the same pathways, could impair the immune response to vaccination or infection [9,10]. Accordingly, the effectiveness of BCG vaccination for tuberculosis is reported to increase with increasing latitude (lower UVR) [11].
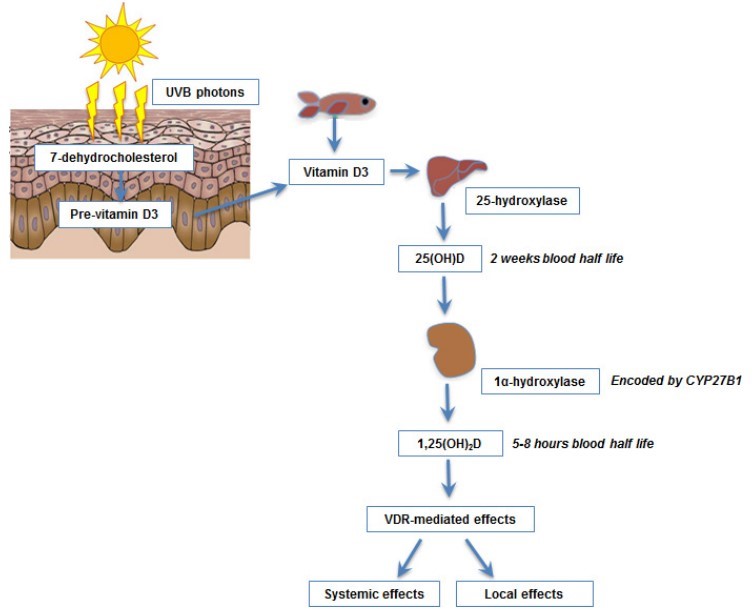
Fig 1
UVB photons are absorbed by 7-dehydrocholestrol in the epidermis and are converted into pre-vitamin D which undergoes a thermal isomerisation to formvitamin D. This then undergoes two hydroxylation reactions, first in the liver to form 25-hydroxyvitamin D (25(OH)D) and then in the kidney to form the active metabolite, 1,25-dihydroxyvitamin D (1,25(OH)2D). Serum 25(OH)D levels are used to determine vitamin D status. VDR, vitamin D receptor.
Observational studies have, in general, supported these ecological patterns. In case-control studies, participants with one of these autoimmune diseases tend to report lower past sun exposure, and/or have lower vitamin D status (measured as the blood concentration of the intermediary metabolite, 25(OH)D, see Figure 1), than healthy controls (see for example recent reviews [12,13]). However, for at least some of the immune-related diseases, it is difficult to determine whether low sun exposure or vitamin D cause, or are caused by, the disease. There is a smaller body of evidence from prospective cohort studies, in particular, because these diseases caused by immune dysfunction are uncommon and require large numbers of participants to be under observation for a sufficient time to achieve required sample sizes. This is a stronger study design because the direction of causality is established, that is, the low vitamin D status precedes the onset of the health outcome. Lower vitamin D status has been linked to increased risk of multiple sclerosis (see case study [14]) and to type 1 diabetes [15], although not all studies show a protective association [16].
Despite these relatively coherent findings from ecologic and observational studies, and plausible biological mechanisms, trials of vitamin D supplementation for the prevention of immune-related diseases have largely returned null results [17]. It is worth considering the challenges of a true prevention trial for these diseases, they are uncommon, possibly with long subclinical phases, with the optimum time for intervention unknown, amongst other difficulties. Prevention trials to date have focused on high-risk populations where the disease process may already be underway, or on preventing progression in those who have already developed the disease, where different pathological processes may be operating (see comment in [18]). Nevertheless, there is not yet compelling evidence from these studies that vitamin D supplementation ameliorates any immune-related disease.
Mechanisms by which l,25(OH)2D may regulate immune cell function
It is generally believed that all immune cell types can respond to 1,25(OH)2D. This belief is driven by recognition of cellular expression of the vitamin D receptor (VDR), the finding of multiple primary 1,25(OH)2D target genes in immune cells and the discovery that many immune cells (macrophages, dendritic cells, T and B lymphocytes) can convert 25(OH)D to 1,25(OH)2D through CYP27B1 activity and therefore provide significant local levels of 1,25(OH)2D for functional outcomes (for review [8,19]). The amount of 1,25(OH)2D produced may depend on the ability of immune cells to express CYP27B1 and other enzymatic machinery of the vitamin D pathway, including the CYP24A1 deactivation enzyme. As an example, in vitro stimulated macrophages produced more 1,25(OH)2D than dendritic cells, which expressed truncated CYP27B1 transcripts resulting in lower CYP27B1 protein levels, and also expressed increased levels of CYP24A1 mRNA [20]. Despite in vitro studies reporting significant biological effects of 1,25(OH)2D on immune cells (frequently under ideal conditions using potentially supra-physiological concentrations of 1,25(OH)2D), the question remains as to the real biological effects of 1,25(OH)2D in vivo. The issue is further complicated by the finding that human cells are frequently used for in vitro investigations, but the majority of studies reporting the biological effects of 1,25(OH)2D in vivo have been performed in the experimental mouse. There are also differences between the regulatory potential of supplemented levels versus homeostatic concentrations of 1,25(OH)2D. An extreme is seen in mice not expressing the VDR; they have increased sensitivity to autoimmune diseases [21] but this finding contributes little information to the extent by which 1,25(OH)2D may dose-dependently regulate VDR-expressing cells. Further work is required to better understand the regulatory properties of 1,25(OH)2D because, as summarised below, many of the findings in vitro have not been replicated in vivo. A central concept is that 1,25(OH)2D stimulates innate immunity and suppresses adaptive immunity, but the mechanisms by which these outcomes are achieved vary with different experimental models.
Exposure of differentiating dendritic cells to 1,25(OH)2D prevents their full maturation [22]. However, debate remains surrounding the properties of these "tolerogenic" dendritic cells and their ability to interfere with T cell division and promotion of T regulatory cell (TReg) production and expansion [23]. Some studies suggest that existing CD25+Foxp3+TReg cells proliferate rather than TReg cell development occurring de novo [24]. Other investigators report that, whilst high non-physiological concentrations of 1,25(OH)2D (10-6M) and long culture periods (2 weeks) are required for induction of Foxp3+ TReg populations, the frequency in vitro of Foxp3+ TRegs can be increased by supplementation of cells with TGF(3 (2 ng/ml) and lower concentrations of 1,25(OH)2D (10-7M) [25]. Upon 1,25(OH)2D administration in vivo, the properties of the exposed dendritic cells are varied. In one model, 1,25(OH)2D was applied topically to the skin of mice [26]. After 18 hours, when the dendritic cells would have migrated to the draining nodes, the capacity of these dendritic cells to take up, process and present antigen to co-cultured T cells was not modified. However, upon transfer to new naive mice, these dendritic cells induced significantly smaller ear-swelling responses, as a measure of contact hypersensitivity. The dendritic cells from the mice treated with topical 1,25(OH)2D expressed increased levels of indoleamine 2,3-dioxygenase which may explain this altered dendritic cell property in vivo [26].
T cells may also directly respond to 1,25(OH)2D. Naive T cells express low levels of the VDR that is up-regulated by antigen-specific triggering of T cell receptors and contributes to priming of naive cells (for review [27]). As VDR expression can also inhibit the transcription of the interleukin (IL)-2 gene, this may represent another point of immune regulation by 1,25(OH)2D. Homing receptors on T cells may be altered by 1,25(OH)2D [28] and a recent analysis of the impact of 1,25(OH)2D, given by oral gavage to mice immunised with myelin oligodendrocyte glycoprotein to induce experimental autoimmune encephalomyelitis, suggests that this may be one of the most important immunoregulatory roles of 1,25 (OH)2D [29]. These authors found that 1,25(OH)2D prevented accumulation of inflammatory cells into the central nervous system, although 1,25(OH)2D did not affect the activation of the pathogenic interferon-g and IL-17-producing T cells in lymph nodes, spleen or the immunisation site, that is, the dramatic systemic immune reaction to myelin oligodendrocyte glycoprotein was not altered. There was no induction of Foxp3+ TReg by 1,25(OH)2D. More specifically, 1,25(OH)2D seemed to maintain the activated Th1/Th17 cells in the circulation and prevented them from crossing the blood brain barrier. The mechanism proposed was a significant and reversible reduction in expression of the chemokine receptor, CXCR3 [29]. Modulation of other chemokine receptors onT cells by 1,25(OH)2D has also been reported [30]. Further, 1,25(OH)2D can reduce the migration of macrophages by suppressing the expression of CCR2, the receptor for monocyte chemotactic protein-1, principally by reducing endoplasmic reticulum stress [31].
TRegs may also be direct targets of 1,25(OH)2D. Using topical delivery to shaved skin, 1,25(OH)2D enhanced the ability of CD4+CD25+ cells in the skin draining lymph nodes to down-regulate T helper type 2-driven asthmatic responses, upon transfer to allergen-sensitised mice [32]. Further investigations found that 1,25(OH)2D, in the presence ofIL-2, directly enhanced the regulatory potential of CD4+CD25+ T cells to control immune responses [33].
Circulating levels of 1,25(OH)2D are a log fold lower than those of 25(OH)D and are not sufficient for immunor- egulation; it is generally proposed that 25(OH)D is converted locally to sufficient levels of 1,25(OH)2D to achieve biological activity. Mast cells have recently been identified, with macrophages, dendritic cells and T and B lymphocytes, to express CYP27B1 for local 1,25(OH)2D production [34]. However, the level of vitamin D binding protein, and its affinity to 25(OH)D, can also restrict the availability of 25(OH)D to dendritic cells and possibly other cells [35]. Cells from individuals with various vitamin D-binding protein (VDBP) variants were studied and for individuals with the strong 25(OH)D binding variant, the availability of 25(OH)D to dendritic cells was restricted, resulting in less dendritic cell-T cell interaction. In summary, we still have much to learn about the mechanisms by which 1,25(OH)2D may regulate immune cell activity in vivo.
Multiple sclerosis: a case study
One of the earliest indications of an association between vitamin D and disorders of human immune function was the geographical variation in prevalence of multiple sclerosis, as first described in 1922 [36]. Additional studies since then have confirmed a latitudinal gradient, with higher incidence or prevalence at locations further from the equator in both the northern and southern hemispheres [1,37,38]. Building on these ecological studies, observational studies confirmed increased risk of multiple sclerosis in association with lower sun exposure [39,40] and with lower 25(OH)D levels [14,41]. Yet, despite plausible immunological pathways whereby vitamin D (through stimulation of TRegs and dampening of T helper type 1 over-reactivity) could diminish the risk of multiple sclerosis or disease activity, vitamin D supplementation trials have shown immunological [42] and radiological [43], but not clinical, benefit for people with multiple sclerosis.
It is challenging to differentiate between vitamin D-dependent and -independent pathways in the effect of sun exposure on disease risk. Limited success in vitamin D supplementation trials suggests that vitamin D- independent effects may contribute more strongly to disease risk. In the mouse, the onset of experimental autoimmune encephalomyelitis, a model for multiple sclerosis, was significantly delayed following chronic irradiation with sub- erythemal doses of UV-B radiation [44]. Serum 25(OH)D levels were only slightly elevated and suppression of disease did not occur following oral administration of 25(OH)D [44]. However, when 1,25(OH)2D was administered (in food), it suppressed the development of experimental autoimmune encephalomyelitis, but only when given at levels that caused hypercalcaemia; one hypothesis was that it was hypercalcaemia per se that was important to disease suppression. TReg-inducing tolerogenic dendritic cells, induced by UV-B irradiation of the skin, were required for amelioration of experimental autoimmune encephalomyelitis [45]. Serum 25(OH)D levels and sun exposure have been shown to be independently associated with the onset of central nervous system demyelination in humans [41] and brain and spinal cord lesions detected by magnetic resonance imaging in people with multiple sclerosis [46]. Additionally, while increased sun exposure during childhood was inversely associated with risk of multiple sclerosis, there was no decreased risk associated with intake of vitamin D supplements during childhood [40].
Sun exposure during childhood may be a more significant factor for disease risk than sun exposure in adulthood. On the basis of migration studies, the risk of developing multiple sclerosis may already have been determined before the age of 15 years [47]. Migrants moving from a country with high prevalence of multiple sclerosis to a country with a lower prevalence retain the high risk of their country of origin if migration occurs after the age of 15, but have the lower risk of their new country if they migrate before 15 years of age [47,48]. However, sun exposure even earlier than childhood may be an important factor in risk of multiple sclerosis. An association between month of birth and risk of multiple sclerosis has been observed in both the northern [49,50] and southern [51] hemispheres, suggesting an important role for sun exposure in utero for the risk of developing the disease in later life. In the experimental autoimmune encephalomyelitis animal model, vitamin D supplementation in the postnatal period delays the onset of the disease [52] but there is no evidence that antenatal vitamin D supplementation has any effect. Although childhood vitamin D levels and sun exposure appear to be important determinants of the risk of disease onset, higher 25(OH)D levels early in the disease course may predict the rate of disease progression in multiple sclerosis [53].
An indirect pathway: vitamin D, immunity and the microbiome
Following interactions with pathogens, immune cells like monocytes can synthesise 1,25(OH)2D, possibly through activation of specialised immune receptors like the toll-like receptors [54], or following stimulation with other immune-modulators like TGF(3 or interferon-g [55]. This stimulates autophagy (the degradation of internal cell structures) and the synthesis of antimicrobials, such as cathelicidin, for bacterial killing [54]. Antimicrobial proteins are expressed at common epithelial surfaces, including the gut, skin, urinary tract and lung [55]. Cathelicidins target gram-positive and -negative bacteria, viruses and fungi by membrane disruption, and are expressed by neutrophils, macrophages, natural killer cells, mast cells and epithelial cells [56,57]. They are widely conserved in mammals - the LL-37 peptide in humans and cathelin- related antimicrobial protein in mice. Transcription of the CAMP, the gene encoding cathelicidin, is regulated by the vitamin D receptor through 1,25(OH)2D in humans, but not rodents [58]. Induced antimicrobials such as cathelicidin and E-defensin-4 [54], can also act as immune-modulators [57]. This pathway (Figure 2) may be dependent upon levels of circulating 25(OH)D, the substrate for 1,25(OH)2D, or of CYP27B1, the enzyme controlling this reaction; however, in vivo evidence for this is lacking. Vitamin D supplementation increases serum levels of 25(OH)D and cathelicidin [58-60], suggesting that systemic elimination of pathogenic microbes is possible; but, supplementation trials aiming to control infections with Mycobacterium tuberculosis have not been successful [61]. In vitro bacterial targets of vitamin D include M. tuberculosis, Pseudomonas aeruginosa and other species [54], and there are reported benefits of vitamin D for the control of viral or fungal infections, especially those of the lower respiratory tract like influenza A [8].
The microbiome constitutes the commensal bacteria that colonise various parts of the body like the gastrointestinal [62] and respiratory [63] tracts and skin [64]. In murine models, 1,25(OH)2D may be able to suppress tissue inflammation by altering the microbiome [65, 66]. Vitamin D deficiency increased the severity of colitis and bacterial numbers in the colons of mice [66] and, in male mice with allergic airway disease (modelling allergic asthma), vitamin D deficiency increased lung inflammation and bacterial numbers [65]; these effects were reversed by vitamin D supplementation [65]. Other studies have linked vitamin D with specific changes to the composition of the bacterial flora in the gut microbiome. The absence of the 1a-hydroxylase enzyme in CYP27B1-/- mice with colitis increased the burden of the Proteobacterium phylum (including Helicobaceteraceae species) in faeces [67]. These observations were linked with diminished numbers of tolerogenic CD103+ dendritic cells in the lamina propria, cells that shape the TReg cell repertoire of the gut [68]. Treatment of these mice with 1,25(OH)2D (1.25 mg/100g diet) suppressed colitis severity and Helicobaceteraceae numbers. Similar observations have been made in humans, whereby increased dietary vitamin D changed the composition of faecal microbiota [69]. Collectively, these studies suggest that vitamin D is instrumental in regulating the microbiome of the lungs and gut, and is entwined with the maintenance of immune tolerance.
As murine studies are starting to illustrate in the gut and lungs, the interplay between vitamin D, the microbiome and immune tolerance may also curb skin inflammation. Abnormal microbiomes have been detected in diseased skin, including psoriatic plaques and atopic dermatitis [70]. Topical 1,25(OH)2D or related analogues are used to treat psoriasis and may be useful for treating atopic dermatitis and other inflammatory skin diseases [71]. The mechanisms by which 1,25(OH)2D suppresses skin inflammation associated with these diseases may at least partially involve promotion of immune tolerance with the activation of TRegs [72]. An added complexity in psoriasis is the levels of cathelicidins that accumulate in the dermis, which can have pro-inflammatory effects at high concentrations [73]. We anticipate that a deeper understanding of how vitamin D modulates the micro- biomes of the skin and other tissues will come through ongoing vitamin D supplementation trials [74].
Other studies suggest that the microbiome may modify the capacity of tissue regulation by vitamin D. In rodent models, probiotics can upregulate expression of the VDR in the colon, reducing inflammation and delaying the transition to dysplasia and cancer [75]. Furthermore, vitamin D supplementation may suppress the development of intestinal tumours in susceptible mice [76]. In a clinical setting, expression of the VDR was decreased in patients with dysplasia and colitis-associated colorectal cancer [77], suggesting that inflammation may also downregulate the VDR. These data suggest that not only can vitamin D modulate tissue inflammation through modifications to the microbiome, but the reverse is also possible—the microbiome and inflammation can change the responsiveness of tissues to vitamin D through regulation of the VDR. 1,25(OH)2D also upregulates the VDR [78], suggesting that this reduced responsiveness may be reversed by supplementation.
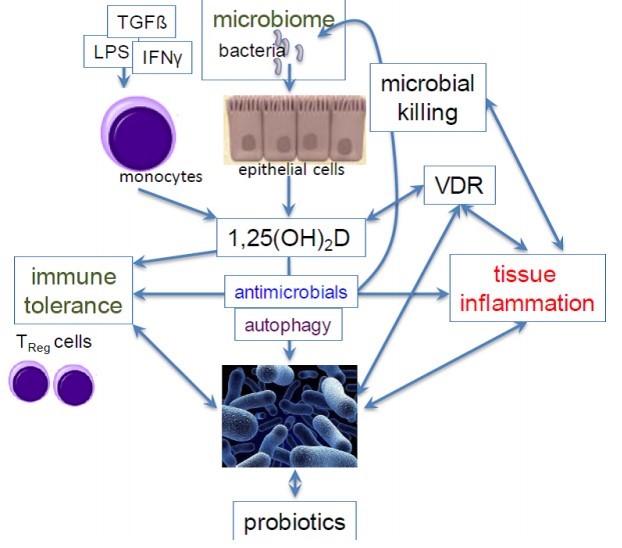
Figure 2.
A proposed network of interactions between vitamin D and the microbiome that may sway the development of immune tolerance or tissue inflammation
Activation of immune cells like monocytes and epithelial cells by bacterial-derived products (e.g. lipopolysaccharide [LPS], derived from the outer membrane of gram-negative bacteria) and cytokines (such as transforming growth factor-3 [TGF3] and interferon-g [IFNg], results in local synthesis of l,25(OH D, and immune tolerance through effects on T eg cells. l,25(OH)2D may also have effects on innate pathways including synthesis of antimicrobials and activation of autophagy, which together may modulate the local microbiome. Alternatively, microbiome changes, like those induced followingthe use of probiotics, may regulate the responsiveness of immune cells to vitamin D by enhancing the expression of the vitamin D receptor (VDR) thus reducing tissue inflammation.
Why doesn’t vitamin D supplementation decrease disease risks?
In the face of supportive observational and mechanistic evidence, why doesn't vitamin D supplementation decrease disease risks? A broad range of responses have been proposed to answer this question with a particular focus on reverse causality or confounding. For the former, it has been proposed that inflammation causes depression of 25(OH)D levels [17] and this accounts for the association between low 25(OH)D levels and increased disease risk. For evidence arising from case- control studies, where disease has already occurred in the cases, this is certainly plausible; however, in the published cohort studies that show a link between low 25(OH)D and development of disease, the latter often occurs many years after the blood sample was taken (e.g. [14]). That asymptomatic disease of sufficient severity to lower 25(OH)D levels was present at this much earlier time seems unlikely. Residual confounding from physical activity or a generally healthier lifestyle may provide an explanation for the significant associations seen in observational studies for some disease outcomes, but neither of these is a known risk factor for autoimmune diseases (and thus could not be a confounder). In addition, with respect to an ability of supplemental vitamin D to alter the course of autoimmune diseases, there is debate about the validity of biochemical or cellular markers from blood as truly representative of disease status.
There are several potential other explanations that need to be considered and each has important implications for research and clinical practice.
Independent beneficial effects of sun exposure
Exposure to UVR and to 1,25(OH)2D both cause immune suppression that is relevant to some (Th-1) autoimmune diseases [8]. Low levels of 25(OH)D seen in observational studies may be a marker for low levels of sun exposure, with the latter also important for disease risk. Vitamin D supplementation studies take account only of vitamin D, without the possibly necessary additional benefits of sun exposure.
Importance of the life stage
There is some evidence to support the importance of early life exposures in the risk of autoimmune diseases. It is challenging to adequately study these exposures. However, several studies suggest that low vitamin D status or low levels of ambient UVR during the in utero period [49], or during childhood [40], are associated with increased risk of immune-related disorders. The lack of any protective effect of vitamin D supplementation in adulthood may be because the processes necessary for disease have already been put in place much earlier in life.
Genetic variation in the physiological response to vitamin D supplementation
A high degree of individual variability in the 25(OH)D response to vitamin D supplementation has been described that is the result ofdemographic, environmental and genetic factors [79]. The effect of these factors on the physiological, or disease-relevant, response to vitamin D supplementation has not been explored.
Corruption of the vitamin D system by the disease
Mismanagement of the vitamin D system may arise during some pathological processes. For example, in leprosy, the Mycobacterium leprae may hijack the use of cellular microRNAs to block vitamin D-induced antimicrobial production [80]. In addition, in some granulomatous diseases, such as sarcoidosis, CYP27B1 is over-expressed in disease-activated macrophages [81] causing high levels of 1,25(OH)2D and increased risk of hypercalcemia.
Free versus total serum 25(OH)D
Vitamin D metabolites in blood are largely tightly bound to VDBP, with a small proportion of the total concentration loosely bound to albumin or unbound (free) [82]. Traditionally the level of total 25(OH)D has been considered the best estimate of vitamin D status, but recent studies have suggested that the "bioavailable" (albumin-bound and free) 25(OH)D concentration may be more relevant to health and disease [83]. The ratio of total to bioavailable 25(OH)D depends on several VDBP- related genes and health states, such as liver disease [84], type 1 diabetes [85], and pregnancy [86].
Vitamin D2 versus vitamin D3 versus active 1,25(OH)2D
There is mixed evidence on the different physiological potency of vitamin D3 compared to D2 (for example, [87,88]). In most observational studies that have separately quantified 25(OH)D2 and 25(OH)D3, levels of the former are generally low or undetectable [89]. Most recent vitamin D supplementation trials use vitamin D3, suggesting that any differences between D2 and D3 do not account for the different findings from observational and vitamin D supplementation trials. The active form, 1,25(OH)2D, is seldom used in supplementation trials. It has a short half-life in blood, can cause hypercalcaemia and raising 1,25(OH)2D levels in blood initiates its deactivation by the 24-hydroxlyase enzyme [90,91].
The study participants were not vitamin D deficient at baseline
If there is a causal association between vitamin D and disease risks, increased risk occurs in association with vitamin D deficiency [92]. Vitamin D supplementation trials commonly include participants who do not have low baseline 25(OH)D levels and whose disease risk would thus be unlikely to change much [91]. Future work could focus on supplementation trials including only people with low baseline levels of 25(OH)D.
Risks to supplementing people who are already vitamin D sufficient
Several studies have now reported U-shaped or reverse J-shaped curves for a range of disease outcomes; for example, for all-cause mortality, both high and low 25(OH)D levels are associated with increased risk [92]. Similarly, in one RCT of high-dose vitamin D supplementation there was an increased risk of falls and fractures [93]. It is likely that there is a loss of study power and a risk of harm if participants in vitamin D supplementation trials already have adequate 25(OH)D levels at baseline [91].
Higher 25(OH)D levels are not maintained over a disease-relevant time period
Many of the diseases for which increased risk has been linked to vitamin D deficiency develop over a long period of time, for example, cancers, and cardiovascular disease. Yet vitamin D supplementation trials are generally relatively short-term, with supplementation for only 1-2 years. This discrepancy in timing may contribute to the failure of vitamin D supplementation trials to decrease the risk of disease onset.
Conclusion
The jury is still out on the importance of vitamin D for immune function: both for vitamin D deficiency increasing the risk of autoimmune diseases and viral infections through loss of regulatory adaptive immune functions, as well as for high vitamin D levels decreasing the risks of tuberculosis and infections with other intracellular pathogens through upregulation of innate immunity. Effects may not be direct, but could work through alterations in the microbiome, or be only partly vitamin D-mediated, with exposure to UVR itself also important. Many of the outstanding questions will be difficult or impossible to resolve in human studies, such as the importance of the timing of any intervention. Pragmatic solutions such as moving whole population distributions of 25(OH)D levels to above a minimum of 40-50 nmol/L may be required, with any beneficial effects only apparent retrospectively.
Abbreviations
1,25(OH)2D, 1,25 dihydroxyvitamin D, the active form of vitamin D; 25(OH)D, 25-hydroxyvitamin D, an intermediate metabolite of vitamin D, the serum concentration is the usual measure of vitamin D status; BCG, Bacillus Calmette-Guerin vaccination for tuberculosis; CCR2, the receptor for monocyte chemotactic protein-1; CD4+CD25+, a type of regulatory T cell; CD25+Foxp3+ TReg, specific type of regulatory T cell; CYP24A1, the gene encoding the 24-hydroxylase enzyme that converts 1,25(OH)2D to 24,25(OH)2D; CYP27B1, the gene encoding for the 1a-hydroxylase enzyme that converts 25(OH)D to 1,25(OH)2D; CXCR3, chemokine receptor 3; DC, dendritic cells; EAE, experimental autoimmune encephalomyelitis, the animal model for MS; IL, interleukin; LL-37, a cathelicidin; LPS, lipopolysaccharide; MS, multiple sclerosis; TGF(3, transforming growth factor beta; Th1 cells, T helper 1 cells; Th17 cells, T helper 17 cells; TReg cell, regulatory T cell; UV-B, shorter wavelength (280-315 nm) ultraviolet radiation; UVR, ultraviolet radiation; VDBP, vitamin D-binding protein; VDR, vitamin D receptor.
Disclosures: The authors declare that they have no disclosures.
References in PDF
See also VitaminDWiki
Gut Microbiota: The Neglected Endocrine Organ (vitamin D not mentioned) – July 2014
Innate and adaptive immune systems probably helped by vitamin D – Oct 2014
Probiotics, prebiotics, and the host microbiome - the science of translation – June 2013
Search VitaminDWiki for Robyn Lucas - one of the authors 29 items as of April 2015
The sun provides more health benefit than vitamin D – Dr. Lucas podcast – May 2015
Antibiotics and Vitamin D are associated with many of the same diseases
Huge increases in health problems – risk factors include Vitamin D, Antibiotics, and Roundup
Exploring gut microbes in Human health and disease: pushing the envelope – Aug 2014
Leprosy associated with changes to Vitamin D receptor genes (VDR)– April 2015