Vitamin D is linked to metabolic syndrome and obesity
Linking the metabolic syndrome and obesity with vitamin D status: risks and opportunities for improving cardiometabolic health and well-being
Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy,http://doi.org/10.2147/DMSO.S176933
Meis Moukayed1 William B Grant2
'School of Arts and Sciences, American University in Dubai, Dubai, UAE; 2 Sunlight, Nutrition, and Health Research Center, San Francisco, CA 94164-1603, USA
Correspondence:William B Grant Sunlight, Nutrition, and Health Research Center, PO. Box 641603, San Francisco, CA 94164-1603, USA Email wbgrant@infionline.net
📄 Download the PDF from VitaminDWiki
The global death toll from noncommunicable diseases is exceptionally high, reported to cause 71% of global deaths worldwide. Metabolic syndrome risk factors, especially excessive adiposity and obesity, are at the heart of the problem resulting in increased comorbidities such as cardiometabolic diseases and cancer, increased health costs, poorer quality of life, and shortened survival. Vitamin D3 can positively reverse many of these adverse effects and outcomes through blocking signaling mechanisms that predispose to cardiometabolic and metastatic disease. As an affordable natural agent, vitamin D3 can be used to counteract obesity-induced inflammation, block early adipogenesis, enhance glucose uptake, counteract hyperleptinemia, ameliorate insulin resistance, and reduce hypertension. This is supported by data from in vitro, in vivo and epidemiological studies and clinical trials. We propose that everyone in general and obese patients in particular consider raising 25-hydroxy’vitamin D levels through UVB exposure and/or supplemental vitamin D3 intake to reduce cardiometabolic and metastatic disease and increase longevity.
Introduction
The metabolic syndrome is characterized by a number of inter-dependent risk factors or diseases that collectively increase a person’s risk for developing type 2 diabetes mellitus (T2DM) and cardiovascular diseases (CVDs). These risk factors include elevated blood glucose levels, hypertension, dyslipidemia (specifically increased low-density lipoprotein (LDL) cholesterol, reduced high-density lipoprotein (HDL) cholesterol, and elevated triglycerides (TG)), increased fat mass and adiposity with intraabdominal fat accumulation, a large waist circumference and high waist to hip ratio. All of the above abnormalities lead to obesity, which is characterized by high body mass index (BMI) >30 kg/m2 and increased adiposity. Obesity is also an independent risk factor of the metabolic syndrome and cardiometabolic diseases.1 Obesity is thought to double the risk of co-morbidity and mortality from noncommunicable diseases (NCD) and their complications, including T2DM, CVDs, and cancer.2,3 This indicates a complex relationship among obesity, cardiometabolic diseases, T2DM, and cancer,4 suggesting that many mechanistic regulatory pathways are common, perhaps intersecting, and may be collectively modulated in our search for therapeutic solutions. At the intersection of all these diseases is increased adiposity and inflammation associated with increasing obesity levels.5
Global obesity rates remain unacceptably high. The WHO estimates of 2016 indicated that over 1.9 billion adults were overweight and over 650 million were obese. Unfortunately, childhood obesity is also on the rise globally, with over 41 million children under the age of 5 years characterized as overweight or obese and 18% of children aged 4-18 years as obese.6 The global NCD burden remains shockingly high and is responsible for 71% of deaths worldwide.7 The cost of co-morbidities, loss of productivity, increases in associated conditions such as mental health decline and poor quality-of-life associated with rising incidence of obesity and metabolic syndrome will greatly challenge global health services worldwide. The absolute global economic burden of these diseases is predicted to increase to $2.1 trillion by 2030.8,9 Therefore, a global drive by all health stakeholders, including countries, global health institutions, health authorities, and private stakeholders, must be coordinated to reduce disease morbidity, mortality, and cost burdens. This can in part be achieved through implementing cost-effective interventions, such as education enabling basic exercise or nutrition interventions to prevent or reduce the collective disease burden.10 These can include increased exercise, encouragement of weight loss, or dietary management.1 The latter could include supplementation with natural agents that can positively affect health, either through preventing risk factor occurrence, disease onset or pathological complications, via acting on several potentially intersecting metabolic pathways common to obesity, cardiometabolic diseases, and cancer. We propose below an argument for the use of vitamin D3 for reduction of risk in obesity-associated NCD. How can we make this link? Epidemiological studies, which we review below, show that low circulating plasma levels of vitamin D3 are present in obese individuals and are associated with poor morbidity and mortality outcomes from T2DM, CVD, and cancer. These have been shown to be ameliorated or reversed upon vitamin D3 supplementation. Moreover, animal models indicate that vitamin D3 is directly involved or implicated in adipogenesis and differentiation. Finally, mechanistic findings in vitro suggest that vitamin D3 can simultaneously modulate several common intersecting hormone-regulated, obesity-induced, inflammation-potentiated signaling pathways common to T2DM, cardiometabolic diseases, and cancer.2 As it is estimated that
- Treatment of vitamin D deficiency on average costs less than 0.1% of the cost of investigations and treatment of worsening comorbidities and complications associated with hypovitaminosis D (ie, “vitamin D treatment cost of $12/year versus, average cost for managing complications, of $6,000 to 20,000/year per affected person” ).11
Then, vitamin D3 supplementation appears to be a potentially suitable cost-effective preventive and therapeutic agent that should be considered for all individuals, and especially in obese patients or those diagnosed with established metabolic syndrome risk factors to improve health outcomes and survival.
Vitamin D metabolism and genomic action in the human body
Vitamin D refers to a fat-soluble secosteroid which can be consumed in foods like supplemented milk, eggs and fish or synthesized in the body following exposure to UV light. Its effects were first identified by Mellanby and McCollum12,13 in cod liver oil and its biochemical metabolism pathways and physiological mechanisms of action for good health and well-being have been described in many tissue types and systems over the years.14-16 Vitamin D3 is initially synthesized from 7-dehydrocholesterol upon UV exposure, then gets converted, respectively, to 25-hydroxyvitamin D [25(OH)D] in the liver, then the biologically active metabolite 1,25-dihydroxyvitamin D [1,25(OH)2D3] in the kidneys. 25(OH)D is the circulating metabolite which gets measured routines as a marker of vitamin D sufficiency in the blood. Hydroxylation of vitamin D to 1,24,25(OH)2D3 by 25-hydroxyvitamin D 24- hydroxylase (24-OHase), an enzyme found in liver, adipocytes and other cell types, results in the inactivation of vitamin D and its destination for excretion.17 The active metabolite of vitamin D, 1,25(OH)2D3 acts via binding to a member of the nuclear receptor superfamily, namely, the vitamin D receptor (VDR). Upon binding of 1,25(OH)2D3 ligand, VDR translocates to the nucleus, dimerizes with retinoid X receptor (RXR) (or other nuclear factors such as peroxisome proliferator-activated receptors-gamma (PPARy), retinoic acid receptor). The VDR/RXR dimer additionally recruits pioneer transcription factors, such as C/EBPa and SPI1 to activate transcription of several genes in several body organs and systems.14,18,19
Vitamin D and obesity - epidemiological findings
Observational studies have often found an inverse relationship between obesity and serum 25(OH)D concentrations.20,21 The model that seems to best explain the observed correlations is volumetric dilution.22 A study of 686 adults taking <400 IU/d vitamin D found that the best fit to the data was a hyperbolic fit to weight, from 90 nmol/L (36 ng/mL) at 40 kg to 36 nmol/L (14 ng/mL) at 160 kg.22 It is assumed that 25(OH)D is stored in both adipose and non-adipose tissues. A review by Heaney found:
- Mean serum 25(OH)D in all studies combined was 45 nmol/L. At the level of vitamin D repletion represented by this concentration, total body vitamin D would be 14,665 IU for a 70 kg adult woman. 65% of this total was present as native cholecalciferol and 35% as 25(OH) D. Nearly three-quarters of the cholecalciferol was in fat, while 25(OH)D was more evenly distributed throughout the body. (20% in muscle, 30% in serum, 35% in fat, and 15% in all other tissues)23
The same group conducted a 10-week vitamin D supplementation study on 62 obese individuals with mean age ~45 years, weight ~107±20 kg, and BMI ~37±5 kg/m2.24 The changes in 25(OH)D after 21 weeks of supplementation were: for 1000 IU/d, 12±10 ng/mL; for 5000 IU/d, 28 ±10 ng/mL; and for 10,000 IU/d, 51±16 ng/mL. Note that the effect of vitamin D supplementation depended not only on baseline 25(OH)D, vitamin D3 dose, weight, and BMI, but also genetic factors related to the absorption and conversion of vitamin D to 25(OH)D.25
In the VITamin D and OmegA-3 (VITAL) trial, participants in the vitamin D treatment group were given 2000 IU/d vitamin D3.26 Respective baseline and 1 year 25 (OH)D concentrations were 33.3 and 45.9 ng/mL for BMI<25 kg/m2 (n=229), 29.5 and 41.4 ng/mL for 25 kg/m2
A high-dose vitamin D Randomized Controlled Trial (RCT) from Iran on vitamin D-deficient participants found a rate of progression from prediabetes to T2DM that was significantly lower in the intervention group (28% vs 3%; P=0.002).43 A similar trial in the US with mean baseline 25(OH)D of 70 nmol/L did not find that supplementation with 4000 IU/d vitamin D3 for 3.5 years significantly reduced progression from prediabetes to T2DM.44 However, significant reductions in progression were found for those with BMI <30 kg/m [HR=0.71 (95% CI, 0.53-0.95)] and those not taking calcium supplements [HR=0.81 (95% CI, 0.66-0.98)]. There was also a significant reduction in progression for those with baseline 25 (OH)D <30 nmol/L.
A cross-sectional analysis of 6868 adults without T2DM in the National Health and Nutrition Examination Survey examined the relationship between 25(OH)D and IR.45 (IR was defined as the HOMA-IR; >75th percentile, sex-specific: 3.9 in men or 3.6 in women). The analysis found that adding 25(OH)D to the model based on established risk factors could identify an additional 2-4 cases per 1000 people. The general 25(OH)D threshold was 60 nmol/L; however, it varied by ethnicity (Mexican- Americans: 54 nmol/L, non-Hispanic whites: 68 nmol/L, and non-Hispanic blacks: 41 nmol/L). This finding suggests that 25(OH)D is not an important determinant of IR.
A number of RCTs have been conducted to determine whether vitamin D supplementation could reduce IR. One such study was conducted in Melbourne, Australia on 54 overweight or obese adults with a mean age of 32 years who were vitamin D deficient (25(OH)D <50 nmol/L).46 Participants were given a bolus dose of 100,000 IU vitamin D3 or placebo at outset, followed by 4000 IU/d or placebo thereafter for 16 weeks. In the treatment arm, mean 25(OH)D rose from 31 to 88 nmol/L. Differences between the treatment and control arms were not significant for insulin sensitivity or first-phase insulin secretion.
However, an RCT conducted in Iran with 162 participants aged 18-80 years with baseline 25(OH)D =12.5 ng/ mL (31 nmol/L) conducted over a 6-month period did find a reduction in the vitamin D treatment arm.43 Those in the vitamin D treatment arm were given 50,000 IU vitamin D3 pearls once weekly for 3 months, followed by one pearl per month for the next 3 months. 25(OH)D rose from 13 to 41 ng/mL in the treatment arm and from 13 to 16 ng/mL in the control arm. The mean HOMA-IR score dropped from 3.4 to 2.9 and 2.6 after 3 and 6 months, respectively, in the treatment arm and from 3.4 to 3.1 after 3 and 6 months in the control arm, with P=0.04 for the difference between the two arms. Glucose tolerance was not significantly different between the two arms after 3 months (P=0.68) but did reach significance after 6 months (P=0.002). The primary difference between the two studies was duration, but the achieved 25(OH)D was also higher in the Iranian study. These two examples illustrate the problems inherent in conducting vitamin D RCTs.
In vitro, treatment with 1,25(OH)2 D3 improves 2-deoxyglucose uptake in 3T3-L1 adipocytes, implying a positive effect on glucose metabolism. Incubation of cells with 1,25 (OH)2D3 for 24 hrs results in a fourfold increase in insulin- stimulated uptake of 2-deoxyglucose in 3T3-L1 adipocytes compared to controls. Moreover, cells pre-treated with 1,25 (OH)2 D3 and then challenged with pro-inflammatory TNF-a had a 40% higher uptake of 2-deoxyglucose compared to those suppressed in their response to insulin stimulation following TNF-a treatment alone. Treatment with 1,25 (OH)2D3 counteracted the effect of TNF-a and restored glucose uptake response close to normal function (90%). The effect of 1,25(OH)2D3 was mediated by AKT phosphorylation.47 These results have a huge implication for the recommended use of vitamin D3 in diabetic, insulin- resistant and obese patients to improve sensitization to insulin and promote glucose uptake as part of therapeutic protocols.
In an insulin-resistant cell model (IR BeWo model) exhibiting high inflammatory gene expression and elevated mTOR signaling, treatment with 1,25(OH)2VitaminD3 reversed mTOR activation and reduced IR levels and lipid infiltration while decreasing leptin, TNF-a, and IL-6 levels.48
In another diet-induced obesity model, mice fed high levels of vitamin D3, high calcium or high calcium plus vitamin D3 all had lower plasma concentrations of glucose and insulin along with increased levels of adiponectin, the hormone that sensitizes adipocytes to insulin.41 Upon vitamin D3 supplementation, with or without Ca,2+ the decrease in plasma insulin concentrations was almost completely normalized to concentrations measured in controls. Collectively, if animal model findings can be extrapolated to humans, these data may indicate that in vivo dietary supplementation of vitamin D3 may attenuate diet-induced hyperglycemia, increase insulin sensitivity and action, and prevent postprandial hyperinsulinemia if administered to obese individuals. Hyperinsulinemia, present in insulin-resistant, diabetic and obese individuals can pose a secondary health risk by potentiating the incidence of disorders such as polycystic ovary syndrome and cancer, as well as increasing mortality.49
1. Vitamin D’s effect on obesity- induced inflammation
Obesity is associated with increased inflammation.50 Adipocytes can secrete IL-6, which is a major inflammatory mediator in obesity. IL-6 also downregulates adiponectin, an anti-inflammatory adipokine. Decreased adiponectin is associated with increased pro-inflammatory effects associated with NF-kB activation.51 Increased IL-6 concentrations and inflammation are associated with IR and a risk of T2DM and CVD.
In vitro studies show that treatment of lipopolysaccharide (LPS)-stimulated mouse 3T3-L1 adipocytes with 1,25 (OH)2D3 for 24 hrs significantly decreased IL-6 production but had no effect on IL-10 production. Similarly, a reduction in IL-6 production in epididymal WAT was observed in mice fed high-fat diets supplemented with vitamin D3 compared to controls. The in vivo findings in mice are especially promising, as they indicate that vitamin D3 can be considered as an adjuvant to reduce inflammation in obese subjects.52 Similarly, in human adipocytes from two different sources - bone marrow-derived human mesenchymal stromal cells (hMSCs) differentiated into adipocytes and adipocytes isolated from biopsies stimulated with LPS - 24-hr treatment with 1,25(OH)2 D3 resulted in a significant reduction in IL-6 secretion. This was associated with a reduction in nuclear factor-KB (NF-kB) nuclear translocation and activity.53
Treatment with 1,25(OH)2 D3 can reduce pro-inflammatory effects induced by TNF-a in 3T3-L1 adipocytes and adipocyte-macrophage co-cultures. This attenuation of the inflammatory response is mediated by dephosphorylation of mitogen activated protein kinase p38, downregulation of NF-kB, and induction of dual-specificity phosphatases (DUSP) DUSP 1, 10, 11 and 16.47
Human adipocytes pre-sensitized with IL-1P and treated with 1,25(OH)2D3 showed a significant reduction in mRNA levels of pro-inflammatory interleukins IL-6, IL-8 and monocyte chemoattractant protein-1 (MCP-1), along with reduced IL-8 secretion. However, a similar reduction was not observed in adipose tissue biopsies removed from obese subjects receiving an oral treatment of vitamin D3 (7000 IU).54
Observational studies do not lend compelling evidence to support an effect of vitamin D on inflammatory markers
in metabolic syndrome patients. There is only weak evidence from observational studies to show that vitamin D reduces biomarkers of inflammation. For example, an RCT involving overweight/obese adults given high-dose vitamin D for 16 weeks did not find any significant reductions in inflammatory biomarkers.46 However, RCTs involving participants with various diseases have found beneficial effects, such as for T2DM55,56 and polycystic ovary syndrome.57
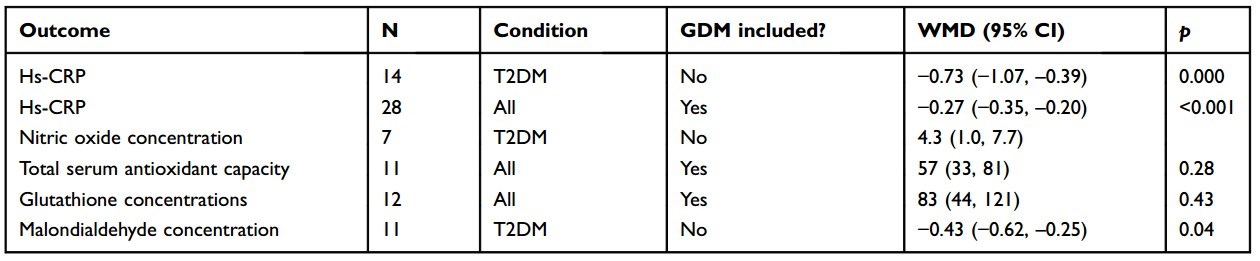
A meta-analysis investigated the effects of vitamin D supplementation on biomarkers of inflammation and oxidative stress in patients with different forms of DM.55 A total of 33 studies conducted between 2012 and 2017 involving 1053 randomized patients were included in the analyses. Twenty of the studies were conducted in Iran, which tended to have patients with low baseline 25(OH)D. Trial durations were from 6 weeks to 12 months. Vitamin D3 doses ranged from 400 IU/d to 50,000 IU every 2 weeks, and some trials included calcium supplements. The effect of vitamin D supplementation on serum high-sensitivity C-reactive protein(hs-CRP) for T2DM was -0.73 (95% CI, -1.07 to -0.39). Most of the other included studies also found significant reductions in hs-CRP, except for studies on pregnant women with gestational DM in People’s Republic of China. Other analyses found significant increases in nitric oxide, antioxidant concentrations, and reduction in malondialdehyde concentration, all in a direction that improves biochemical status (Table 1). However, there was an indication of publication bias for hs-CRP and malon- dialdehyde concentration.
1. Vitamin D regulates circulating lipids, blood pressure and improves CVD outcomes
Table 1 Findings reported in a meta-analysis of the effects of vitamin D supplementation on biomarkers of inflammation and oxidative stress in patients with T2DM55
The endothelium plays an important role in the maintenance of blood fluidity, vessel wall tone and permeability. Endothelial damage via oxidative stress and metabolic disorders contributes to vascular endothelial dysfunction, impaired vascular relaxation, vascular hypertrophy fibrosis, plaque deposition, and arterial stiffness, all of which contribute to hypertension, atherosclerosis and CVDs.58,59 Obesity-associated hyperleptinemia has been shown to cause endothelial cell dysfunction and increase inflammation and oxidative stress in vitro and in vivo.60,61 Obese individuals have been reported to have significantly higher plasma leptin levels compared to eutrophic individuals.62 Most recently, plasma leptin level has been reported to be positively associated with increased blood pressure independent of gender and BMI.63 Recent findings demonstrate that 1,25 (OH)2D3 can counteract leptin-induced oxidative stress and inflammation in human umbilical vein endothelial cells. Treatment of these cells with leptin alone significantly increased reactive oxygen species production, NF-kB translocation to the nucleus, and upregulated the mRNA expression of MCP1, TGFp, and vascular adhesion molecule 1. Pretreatment with 1,25(OH)2D3 at physiological concentrations (10-10 M) and supraphysiological concentrations (10-7 M) prevented leptin-induced superoxide anion production, significantly increased nuclear factor (erythroid-derived 2)- like 2 (NRF2) translocation to the nucleus, and upregulated expression of the antioxidant defense genes superoxide dismutase 2, glutathione peroxidase, NADPH hydrogenase (quinone) 1 (NQO1), and heme oxygenase 1.64 These findings reinforce the idea that 1,25(OH)2D3 may have potent protective effects against oxidative stress-induced damage and hence prevent metabolic complications of obesity such as hypertension or other inflammation-induced complications mediated by increased NFkB activity.
VDR-null mice display less body fat mass and lower plasma triglyceride and cholesterol levels compared with wild-type male mice.38 VDR-/- mice also have higher blood pressure than wild-type mice. These effects are associated with increases in renin (RER) mRNA expression and plasma angiotensin II. These mice remain responsive to challenges in salt variation, though not as effectively as wild-type controls, indicating that VDR plays a role in the renin-angiotensin system and ultimately in blood pressure control. At the gene level, the renin gene RER contains a transcription response element that can be repressed by VDR binding.65,66 Mice with a deletion in VDR also exhibit a reduction in end-diastolic and end-systolic volume, impaired ventricular function and cardiac hypertrophy due to increased calcineurin inhibitory protein 1 expression (MCIP1) and impairments in the calcineurin/nuclear factor of activated T cell (NFATc1) signaling pathways.67
Additionally, studies by Cheng et al indicate that 1,25 (OH)2D3 ameliorates hepatic abnormalities in T2DM. In a mouse model of T2DM, 8-week supplementation of type 2 diabetes db/db mice with 1,25(OH)2D3 ameliorates hepatic triglyceride accumulation and glucose production via the activation of Ca2+ mediated CaMKKp/AMPK signaling pathway.68 Imga et al have shown that vitamin D administration in overweight or obese vitamin D-deficient premenopausal women, can result in a significant reduction in LDL-C levels and improvement of IR.69 Effects on vitamin D on postprandial lipid profiles in obese women may be dose-dependent and favors a reduction in LDL and vLDL at lower vitamin D levels, and an elevation of lipids at higher vitamin D doses.70 Perhaps the latter may be due to the effects of vitamin D on increased lipolysis in adipocytes and P-oxidation associated with concentrated vitamin action on WAT. However, further studies need to be conducted in this respect and should include male subjects to confirm the mechanisms explaining the dose-dependent effects.
As for observational studies, there is limited evidence that vitamin D reduces blood pressure and hypertension. In a 6- month vitamin D supplementation study involving middle- aged African Americans, each 1 ng/mL increase in 25(OH)D resulted in a significant 0.2 mm Hg drop in systolic blood pressure (SBP) but no effect on diastolic blood pressure (DBP).71 In a community-based open-label study in Canada where participants were provided vitamin D3 supplements and encouraged to achieve >40 ng/mL 25(OH)D, those who were hypertensive at baseline (SBP ~155 mm Hg, DBP ~95 mm Hg) reduced their SBP by —14—18 mm Hg and DBP by ~12 mm Hg.72 However, participants who were prehypertensive did not have significant reductions in blood pressure.
However, in CVD outcomes, there is reasonable evidence that vitamin D reduces the risk of CVD. A meta-analysis of 34 prospective observational studies involving 180,667 participants found that the pooled risk ratio for the incidence of a 10-ng/mL increase was 0.90 (95% CI, 0.86-0.94), while the risk ratio for mortality was 0.88 (95% CI, 0.80-0.96).73
A meta-analysis of the effects of vitamin D supplementation on CVD risk factors based on 81 studies found significant beneficial effects for blood pressure, lipid profile, parathyroid hormone, and serum high-sensitivity C- reactive protein (hs-CRP) but not for peak wave velocity or augmentation index.
Additional evidence that vitamin D deficiency plays an important role in the risk of CVD is that CVD rates are 1525% higher in winter than in summer,75 even in warm countries such as Kuwait.76
In general, a majority of the evidence points to a beneficial effect of vitamin D in reducing the risk of CVD, although RCTs are not available to support these associations.77,78 For example, the VITAL study found a HR for major CVD from 2000 IU/d vitamin D3 vs placebo over 5 years =0.97 (95% CI, 0.85—1.12).26 However, VITAL was based on vitamin D dose, while proper RCTs should be based on 25(OH)D.79 A recent metaanalysis of observational studies of CVD events vs 25 (OH)D found few effects above 15 ng/mL.73 The mean baseline 25(OH)D in VITAL was 27.8 ng/mL among 395 males and 31.7 ng/mL in 441 females.26 Thus, a significant beneficial effect would not be expected.
1. Vitamin D reduces risk of and improves survival from cancer
There is very strong evidence that vitamin D reduces the risk of incidence and death for many types of cancer. Geographical ecological studies from mid-latitude countries generally find inverse correlations between indices of solar UVB doses and mortality rates for 15-20 types of cancer.14 Prospective observational studies have found significant inverse correlations between serum 25(OH)D at the time of enrollment and incidence of colorectal cancer.80 Case-control studies have identified a significant inverse correlation between 25(OH)D measured near time of diagnosis, but this correlation was not observed in prospective studies due to the fact that breast cancer can develop rapidly.81 Vitamin D is more effective at reducing cancer mortality rates than incidence rates. For example, a meta-analysis of the effects of vitamin D supplementation on risk of cancer found no significant effect on cancer incidence but a significant 15% reduction in cancer mortality rates.82 The mechanisms whereby vitamin D reduces the risk of cancer are well known. The most important of these affect cellular differentiation, proliferation and apoptosis, angiogenesis, and 14,83,84 metastasis. , , Recently, results of the VITAL trial were released.26 This RCT included over 25,000 participants, including more than 5000 African Americans, who were followed for 5 years. The treatment arm was given 2000 IU/d vitamin D3, and a placebo was given to the control arm. The results revealed three important findings. First, participants with BMI <25 kg/m2 had a HR for cancer incidence =0.76 (95% CI, 0.61-0.90). Second, death from cancer was significantly reduced if results from the first 1 or 2 years were omitted: HR=0.79 (95% CI, 0.63-0.99) for 1 year and 0.75 (95% CI, 0.59-0.96) for 2 years. It is reasonable to perform this type of analysis, as 25(OH)D can affect cancer risk for some time. The third finding was that cancer incidence for black Americans was marginally non-significantly reduced [HR=0.77 (95% CI, 0.59-1.01)]. Other findings regarding vitamin D were not significant. Significant effects on incidence were found among the groups that had lower baseline and/or achieved higher 25(OH)D levels. The main problem with VITAL was that the outcomes were based on vitamin D dose, whereas 25(OH)D should have been used.79
1. Conclusion and recommendations for improved metabolic health and wellbeing
Table 2 Beneficial actions of vitamin D3 bioactive metabolites on various risk factors influencing cardiometabolic health outcomes Factor or marker
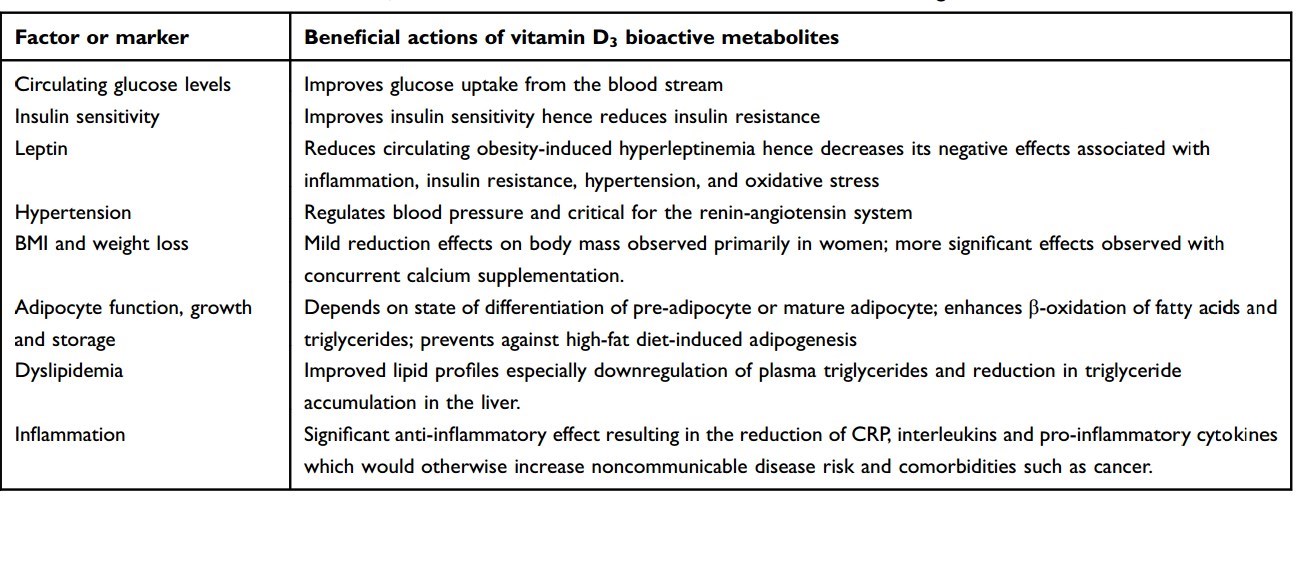
We propose the use of vitamin D3 for all individuals to protect against cardiometabolic and metastatic disease (Table 2). In vitro studies, animal models, observational studies’ findings, and limited number of existing RCTs support the proposal that vitamin D3 will have therapeutic power/benefits to reduce the burden of disease in obese patients by counteracting the effects of many metabolic risk factors. This will greatly improve patient health outcomes, quality of life, and survival. This is expected to reduce health costs of treating NCDs, related comorbidities, and associated indirect loss of productivity costs. Overall, the cost of a maintenance dose of 1000-5000 IU vitamin D3 daily needed to achieve plasma circulating levels in the range of 30-50 ng/mL is cost-effective compared to costs of treatment of NCDs. In addition, there are many additional health benefits of vitamin D supplementation and very limited risk of adverse effects. Given that increased adiposity has the effect of depleting much of the 25(OH)D precursor out of the plasma,85 the higher daily dose of vitamin D3 is advised in obese individuals. More RCTs need to be conducted in obese patients to determine the correct daily dose-titration needed in patients with increasing number of metabolic risk factors or increasing disease severity.
Disclosure Dr William B Grant reports grants from Bio-Tech Pharmacal, Inc., outside the submitted work. The authors report no other conflicts of interest in this work.
1. References
Han TS, Lean ME. A clinical perspective of obesity, metabolic syndrome and cardiovascular disease. JRSM Cardiovasc Dis. 2016;5:2048004016633371. doi:10.1177/2048004016633371
Allott EH, Hursting SD. Obesity and cancer: mechanistic insights from transdisciplinary studies. Endocr Relat Cancer. 2015;22(6):R365- R386. doi:10.1530/ERC-15-0400
Berger NA. Obesity and cancer pathogenesis. Ann N Y Acad Sci. 2014;1311:57-76. doi:10.1111/nyas.12416
Garcia-Jimenez C, Gutierrez-Salmeron M, Chocarro-Calvo A, Garcia- Martinez JM, Castano A, De la Vieja A. From obesity to diabetes and cancer: epidemiological links and role of therapies. Br J Cancer. 2016;114(7):716-722. doi:10.1038/bjc.2016.37
Cifarelli V, Hursting SD. Obesity, diabetes and cancer: a mechanistic perspective. Int J Diabetol Vasc Dis Res. 2015;2015(Suppl 4). doi:10.19070/2328-353X-SI04001
Organization WH. Obesity and overweight factsheets. World Health Organization. Available from: https://www.who.int/news-room/fact- sheets/detail/obesity-and-overweight. Published 2011. Accessed July 16, 2019.
Organization WH. Noncommunicable disease country profile 2018. World Health Organization. Available from: https://www.who.int/ nmh/publications/ncd-profiles-2018/en/. Published 2018. Accessed July 16, 2019.
Bommer C, Sagalova V, Heesemann E, et al. Global economic burden of diabetes in adults: projections from 2015 to 2030. Diabetes Care. 2018;41(5):963-970. doi:10.2337/dc17-1962
Cawley J, Meyerhoefer C. The medical care costs of obesity: an instrumental variables approach. J Health Econ. 2012;31(1):219- 230. doi:10.1016/j.jhealeco.2011.10.003
Bertram MY, Sweeny K, Lauer JA, et al. Investing in non-communicable diseases: an estimation of the return on investment for prevention and treatment services. Lancet. 2018;391(10134):2071-2078. doi:10.1016/S0140-6736(18)30665-2
Wimalawansa SJ. Biology of Vitamin D. J Steroids Horm Sci. 2019;10(1). doi:10.4172/2157-7536.1000198
Mellanby E. An experimental investigation on rickets 1919. Nutrition. 1989;5(2):81—86; discussion 87.
McCollum EV. The paths to the discovery of vitamins A and D. J Nutr. 1967;91(2 Suppl 1):11-16. doi:10.1093/jn/91.2_Suppl.11
Moukayed M, Grant WB. Molecular link between vitamin D and cancer prevention. Nutrients. 2013;5(10):3993 021. doi:10.3390/nu5103993
Baggerly CA, Cuomo RE, French CB, et al. Sunlight and Vitamin D: necessary for public health. J Am Coll Nutr. 2015;34(4):359-365. doi:10.1080/07315724.2015.1039866
Wacker M, Holick MF. Vitamin D - effects on skeletal and extra- skeletal health and the need for supplementation. Nutrients. 2013;5 (1):111—148. doi:10.3390/nu5010111
Dilworth FJ, Scott I, Green A, et al. Different mechanisms of hydroxylation site selection by liver and kidney cytochrome P450 species (CYP27 and CYP24) involved in vitamin D metabolism. J Biol Chem. 1995;270(28):16766-16774. doi:10.1074/jbc.270.28.16766
Carlberg C, Campbell MJ. Vitamin D receptor signaling mechanisms: integrated actions of a well-defined transcription factor. Steroids. 2013;78(2):127-136. doi:10.1016/j.steroids.2012.10.019
Wamberg L, Christiansen T, Paulsen SK, et al. Expression of vitamin D-metabolizing enzymes in human adipose tissue - the effect of obesity and diet-induced weight loss. Int J Obes (Lond). 2013;37 (5):651-657. doi:10.1038/ijo.2012.112
Jungert A, Roth HJ, Neuhauser-Berthold M. Serum 25-hydroxyvita- min D3 and body composition in an elderly cohort from Germany: a cross-sectional study. Nutr Metab (Lond). 2012;9(1):42. doi:10.1186/ 1743-7075-9-42
Lee SH, Kim SM, Park HS, et al. Serum 25-hydroxyvitamin D levels, obesity and the metabolic syndrome among Korean children. Nutr Metab CardiovascDis. 2013;23(8):785-791.doi:10.1016/j.numecd.2012.04.013
Drincic AT, Armas LA, Van Diest EE, Heaney RP. Volumetric dilution, rather than sequestration best explains the low vitamin D status of obesity. Obesity (Silver Spring). 2012;20(7):1444-1448. doi:10.1038/oby.2011.404
Heaney RP, Horst RL, Cullen DM, Armas LA. Vitamin D3 distribution and status in the body. J Am Coll Nutr. 2009;28(3):252-256.
Drincic A, Fuller E, Heaney RP, Armas LA. 25-Hydroxyvitamin D response to graded vitamin D(3) supplementation among obese adults. J Clin Endocrinol Metab. 2013;98(12):4845-4851. doi:10. 1210/jc.2012-4103
Didriksen A, Grimnes G, Hutchinson MS, et al. The serum 25- hydroxyvitamin D response to vitamin D supplementation is related to genetic factors, BMI, and baseline levels. Eur J Endocrinol. 2013;169(5):559-567. doi:10.1530/EJE-13-0233
Manson JE, Cook NR, Lee IM, et al. Vitamin D supplements and prevention of cancer and cardiovascular disease. N Engl J Med. 2019;380(1):33-44. doi:10.1056/NEJMoa1809944
Mawer EB, Backhouse J, Holman CA, Lumb GA, Stanbury SW. The distribution and storage of vitamin D and its metabolites in human tissues. Clin Sci. 1972;43(3):413-431.
Marcotorchino J, Tourniaire F, Landrier JF. Vitamin D, adipose tissue, and obesity. Horm Mol Biol Clin Investig. 2013;15(3):123- 128. doi:10.1515/hmbci-2013-0027
Hoseini R, Damirchi A, Babaei P. Vitamin D increases PPARgamma expression and promotes beneficial effects of physical activity in metabolic syndrome. Nutrition. 2017;36:54-59. doi:10.1016/j.nut.2016.06.010
Babaei P, Shirkouhi SG, Hosseini R, Soltani Tehrani B. Vitamin D is associated with metabotropic but not neurotrophic effects of exercise in ovariectomized rats. Diabetol Metab Syndr. 2017;9:91.doi:10.1186/sl3098-017-0288-z
Pannu PK, Zhao Y, Soares MJ. Reductions in body weight and percent fat mass increase the vitamin D status of obese subjects: a systematic review and metaregression analysis. Nutr Res. 2016;36 (3):201-213. doi:10.1016/j.nutres.2015.11.013
Lupton JR, Faridi KF, Martin SS, et al. Deficient serum 25-hydro- xyvitamin D is associated with an atherogenic lipid profile: the Very Large Database of Lipids (VLDL-3) study. J Clin Lipidol. 2016;10 (1):72—81 e71. doi:10.1016/j.jacl.2015.09.006
Abbas MA. Physiological functions of Vitamin D in adipose tissue. J Steroid Biochem Mol Biol. 2017;165(Pt B):369-381. doi:10.1016/j. jsbmb.2016.08.004
Landrier JF, Karkeni E, Marcotorchino J, Bonnet L, Tourniaire F. Vitamin D modulates adipose tissue biology: possible consequences for obesity? Proc Nutr Soc. 2016;75(1):38 6. doi:10.1017/S0029665115004164
Kong J, Li YC. Molecular mechanism of 1,25-dihydroxyvitamin D3 inhibition of adipogenesis in 3T3-L1 cells. Am J Physiol Endocrinol Metab. 2006;290(5):E916-E924. doi:10.1152/ajpendo.00410.2005
Ricciardi CJ, Bae J, Esposito D, et al. 1,25-Dihydroxyvitamin D3/ vitamin D receptor suppresses brown adipocyte differentiation and mitochondrial respiration. Eur J Nutr. 2015;54(6):1001-1012. doi:10.1007/s00394-014-0778-9
Nobre JL, Lisboa PC, Carvalho JC, et al. Leptin blocks the inhibitory effect of vitamin D on adipogenesis and cell proliferation in 3T3-L1 adipocytes. Gen Comp Endocrinol. 2018;266:1-8. doi:10.1016/j. ygcen.2018.01.014
Wong KE, Szeto FL, Zhang W, et al. Involvement of the vitamin D receptor in energy metabolism: regulation of uncoupling proteins. Am J Physiol Endocrinol Metab. 2009;296(4):E820-E828. doi:10.1152/ ajpendo.90763.2008
Rovner AJ, Stallings VA, Kawchak DA, Schall JI, Ohene-Frempong K, Zemel BS. High risk of vitamin d deficiency in children with sickle cell disease. J Am Diet Assoc. 2008;108(9):1512-1516. doi:10.1016/j.jada.2008.06.433
Sergeev IN. 1,25-Dihydroxyvitamin D3 induces Ca2+-mediated apoptosis in adipocytes via activation of calpain and caspase-12. Biochem Biophys Res Commun. 2009;384(1):18-21. doi:10.1016/j. bbrc.2009.04.078
Sergeev IN. Vitamin D-mediated apoptosis in cancer and obesity. Horm Mol Biol Clin Investig. 2014;20(2):43 9. doi:10.1515/ hmbci-2014-0035
Park SK, Garland CF, Gorham ED, BuDoff L, Barrett-Connor E. Plasma 25-hydroxyvitamin D concentration and risk of type 2 diabetes and pre-diabetes: 12-year cohort study. PLoS One. 2018;13(4): e0193070. doi:10.1371/journal.pone.0193070
Niroomand M, Fotouhi A, Irannejad N, Hosseinpanah F. Does highdose vitamin D supplementation impact insulin resistance and risk of development of diabetes in patients with pre-diabetes? A doubleblind randomized clinical trial. Diabetes Res Clin Pract. 2019;148:1-9. doi:10.1016/j.diabres.2018.12.008
Pittas AG, Dawson-Hughes B, Sheehan P, et al. Vitamin D supplementation and prevention of type 2 diabetes. N Engl J Med. 2019. doi:10.1056/NEJMoa1900906
Al-Khalidi B, Kimball SM, Rotondi MA, Ardern CI. Standardized serum 25-hydroxyvitamin D concentrations are inversely associated with cardiometabolic disease in U.S. adults: a cross-sectional analysis of NHANES, 2001-2010. Nutr J. 2017;16(1):16. doi:10.1186/ s12937-017-0237-6
Mousa A, Naderpoor N, de Courten MP, et al. Vitamin D supplementation has no effect on insulin sensitivity or secretion in vitamin D-deficient, overweight or obese adults: a randomized placebo-controlled trial. Am J Clin Nutr. 2017;105(6):1372-1381. doi:10.3945/ ajcn.117.152736
Marcotorchino J, Gouranton E, Romier B, et al. Vitamin D reduces the inflammatory response and restores glucose uptake in adipocytes. Mol Nutr Food Res. 2012;56(12):1771-1782. doi:10.1002/ mnfr.201200383
Li G, Lin L, Wang YL, Yang H. 1,25(OH)2D3 protects trophoblasts against insulin resistance and inflammation via suppressing mTOR signaling. Reprod Sci. 2019;26(2):223-232. doi:10.1177/19337191 18766253
Tsujimoto T, Kajio H, Sugiyama T. Association between hyperinsu- linemia and increased risk of cancer death in nonobese and obese people: a population-based observational study. Int J Cancer. 2017;141(1):102-111. doi:10.1002/ijc.30729
Karczewski J, Sledzinska E, Baturo A, et al. Obesity and inflammation. Eur CytokineNetw. 2018;29(3):83-94. doi:10.1684/ecn.2018.0415
Lira FS, Rosa JC, Pimentel GD, et al. Both adiponectin and interleukin-10 inhibit LPS-induced activation of the NF-kappaB pathway in 3T3-L1 adipocytes. Cytokine. 2012;57(1):98-106. doi:10.1016/j. cyto.2011.10.001
Lira FS, Rosa JC, Cunha CA, et al. Supplementing alpha-tocopherol (vitamin E) and vitamin D3 in high fat diet decrease IL-6 production in murine epididymal adipose tissue and 3T3-L1 adipocytes following LPS stimulation. Lipids Health Dis. 2011;10:37. doi:10.1186/ 1476-511X-10-232
Mutt SJ, Karhu T, Lehtonen S, et al. Inhibition of cytokine secretion from adipocytes by 1,25-dihydroxyvitamin D(3) via the NF-kappaB pathway. Faseb J. 2012;26(11):4400-4407. doi:10.1096/fj.12-210880
Wamberg L, Cullberg KB, Rejnmark L, Richelsen B, Pedersen SB. Investigations of the anti-inflammatory effects of vitamin D in adipose tissue: results from an in vitro study and a randomized controlled trial. Horm Metab Res. 2013;45(6):456-462. doi:10. 1055/s-0032-1331746
Mansournia MA, Ostadmohammadi V, Doosti-Irani A, et al. The effects of vitamin D supplementation on biomarkers of inflammation and oxidative stress in diabetic patients: a systematic review and meta-analysis of randomized controlled trials. Horm Metab Res. 2018;50(6):429-440. doi:10.1055/a-0630-1303
Yu Y, Tian L, Xiao Y, Huang G, Zhang M. Effect of Vitamin D supplementation on some inflammatory biomarkers in type 2 diabetes mellitus subjects: a systematic review and meta-analysis of randomized controlled trials. Ann Nutr Metab. 2018;73(1):62-73. doi:10.1159/000490358
Akbari M, Ostadmohammadi V, Lankarani KB, et al. The effects of Vitamin D supplementation on biomarkers of inflammation and oxidative stress among women with polycystic ovary syndrome: a systematic review and meta-analysis of randomized controlled trials. Horm Metab Res. 2018;50(4):271-279. doi:10.1055/s-0044- 101355
Dharmashankar K, Widlansky ME. Vascular endothelial function and hypertension: insights and directions. Curr Hypertens Rep. 2010;12 (6):448 55. doi:10.1007/s11906-010-0150-2
Dikalov SI, Dikalova AE. Crosstalk between mitochondrial hyperacetylation and oxidative stress in vascular dysfunction and hypertension. Antioxid Redox Signal. 2019. doi:10.1089/ars.2018.7632
Knudson JD, Dincer UD, Zhang C, et al. Leptin receptors are expressed in coronary arteries, and hyperleptinemia causes significant coronary endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2005;289(1):H48-H56. doi:10.1152/ajpheart.01159.2004
Yamagishi SI, Edelstein D, Du XL, Kaneda Y, Guzman M, Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem. 2001;276(27):25096-25100. doi:10.1074/ jbc.M007383200
Al Maskari MY, Alnaqdy AA. Correlation between serum leptin levels, body mass index and obesity in omanis. Sultan Qaboos Univ Med J. 2006;6(2):27-31.
Ghaedian MM, Nazari Jaz A, Momeni M, Ghaedian T, Samiei N. Plasma leptin level is positively associated with blood pressure measures independent of gender and BMI. Clin Exp Hypertens. 2018;1-5. doi:10.1080/10641963.2018.1557684
Teixeira TM, Da Costa DC, Resende AC, Soulage CO, Bezerra FF, Daleprane JB. Activation of Nrf2-antioxidant signaling by 1,25-dihy- droxycholecalciferol prevents leptin-induced oxidative stress and inflammation in human endothelial cells. J Nutr. 2017;147(4):506- 513. doi:10.3945/jn.116.239475
Demay MB. Physiological insights from the vitamin D receptor knockout mouse. Calcif Tissue Int. 2013;92(2):99-105. doi:10.1007/ s00223-012-9633-2
Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25- Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110(2):229-238. doi:10.1172/JCI15219
Chen G, Kim SH, King AN, et al. CYP24A1 is an independent prognostic marker of survival in patients with lung adenocarcinoma. Clin Cancer Res. 2011;17(4):817-826. doi:10.1158/1078-0432.CCR-10-1789
Cheng S, So WY, Zhang D, Cheng Q, Boucher BJ, Leung PS. Calcitriol reduces hepatic triglyceride accumulation and glucose output through Ca2+/CaMKKp/AMPK activation under insulin-resistant conditions in type 2 diabetes mellitus. Curr Mol Med. 2016;16(8):747-758.
Imga NN, Karci AC, Oztas D, Berker D, Guler S. Effects of vitamin D supplementation on insulin resistance and dyslipidemia in overweight and obese premenopausal women. Arch Med Sci. 2019;15 (3):598-606. doi:10.5114/aoms.2018.75864
Fernandez-Arroyo S, Hernandez-Aguilera A, de Vries MA, et al. Effect of Vitamin D3 on the postprandial lipid profile in obese patients: a non-targeted lipidomics study. Nutrients. 2019;11(5). doi:10.3390/nu11051194
Forman JP, Scott JB, Ng K, et al. Effect of vitamin D supplementation on blood pressure in blacks. Hypertension. 2013;61(4):779-785. doi:10.1161/HYPERTENSIONAHA.111.00659
Mirhosseini N, Vatanparast H, Kimball SM. The association between serum 25(OH)D status and blood pressure in participants of a community-based program taking Vitamin D supplements. Nutrients. 2017;9(11). doi:10.3390/nu9111244
Zhang R, Li B, Gao X, et al. Serum 25-hydroxyvitamin D and the risk of cardiovascular disease: dose-response meta-analysis of prospective studies. Am J Clin Nutr. 2017;105(4):810-819. doi:10.3945/ ajcn.116.140392
Mirhosseini N, Rainsbury J, Kimball SM. Vitamin D supplementation, serum 25(OH)D concentrations and cardiovascular disease risk factors: a systematic review and meta-analysis. Front Cardiovasc Med. 2018;5:87. doi:10.3389/fcvm.2018.00087
Grant WB, Bhattoa HP, Boucher BJ. Seasonal variations of U.S. mortality rates: roles of solar ultraviolet-B doses, vitamin D, gene exp ression, and infections. J Steroid Biochem Mol Biol. 2017;173:5- 12. doi:10.1016/j.jsbmb.2017.01.003
Douglas AS, al-Sayer H, Rawles JM, Allan TM. Seasonality of disease in Kuwait. Lancet. 1991;337(8754):1393-1397. doi:10.1016/0140-6736(91)93069-l
Weyland PG, Grant WB, Howie-Esquivel J. Does sufficient evidence exist to support a causal association between vitamin D status and cardiovascular disease risk? An assessment using Hill’s criteria for causality. Nutrients. 2014;6(9):3403-3430. doi:10.3390/nu6093403
Wimalawansa SJ. Vitamin D and cardiovascular diseases: causality. J Steroid Biochem Mol Biol. 2018;175:29^3. doi:10.1016/j. jsbmb.2016.12.016
Grant WB, Boucher BJ, Bhattoa HP, Lahore H. Why vitamin D clinical trials should be based on 25-hydroxyvitamin D concentrations. J Steroid Biochem Mol Biol. 2018;177:266-269. doi:10.1016/j. jsbmb.2017.08.009
Garland CF, Gorham ED. Dose-response of serum 25-hydroxyvita- min D in association with risk of colorectal cancer: A meta-analysis. J Steroid Biochem Mol Biol. 2017;168:1-8. doi:10.1016/j. jsbmb.2016.12.003
Grant WB. 25-hydroxyvitamin D and breast cancer, colorectal cancer, and colorectal adenomas: case-control versus nested case-control studies. Anticancer Res. 2015;35(2):1153-1160.
Goulao B, Stewart F, Ford JA, MacLennan G, Avenell A. Cancer and vitamin D supplementation: a systematic review and meta-analysis. Am J Clin Nutr. 2018;107(4):652-663. doi:10.1093/ajcn/nqx047
Moukayed M, Grant WB. The roles of UVB and vitamin D in reducing risk of cancer incidence and mortality: a review of the epidemiology, clinical trials, and mechanisms. Rev Endocr Metab Disord. 2017. doi:10.1007/s11154-017-9415-2
Grant WB, Moukayed M. Vitamin D3 from ultraviolet-B exposure or oral intake in relation to cancer incidence and mortality. Curr Nutr Rep. 2019;8(3):203-211. doi:10.1007/s13668-019-0262-5
Wortsman J, Matsuoka LY, Chen TC, Lu Z, Holick MF. Decreased bioavailability of vitamin D in obesity. Am J Clin Nutr. 2000;72 (3):690-693. doi:10.1093/ajcn/72.3.690