Major review of Vitamin D – Holick
The Roles of Vitamin D in Skeletal Muscle: Form, Function, and Metabolism
Endocrine Reviews February 1, 2013 vol. 34 no. 1 33-83
Christian M. Girgis, Roderick J. Clifton-Bligh, Mark W. Hamrick, Michael F. Holick, and Jenny E. Gunton
Garvan Institute of Medical Research (C.M.G., J.E.G.) and St. Vincent's Clinical School (J.E.G.), University of New South Wales, Sydney, New South Wales 2010, Australia; Faculty of Medicine (C.M.G., R.J.C.-B., J.E.G.), University of Sydney, Sydney, New South Wales 2052, Australia; The Kolling Institute of Medical Research (R.J.C.-B.) and Royal North Shore Hospital (R.J.C.-B.), Sydney, New South Wales 2065, Australia; Georgia Health Sciences University (M.W.H.), Augusta, Georgia 30912; Boston University Medical Center (M.F.H.), Boston, Massachusetts 02118; and Department of Endocrinology and Diabetes (J.E.G.), Westmead Hospital, Sydney, New South Wales 2145, Australia
This 56 page study is attached at the bottom of this page includes all graphics and tables
Only a few of which have been extracted below
The study is too long to be translated by Google, so it is in two sections
Recent by Holick in VitaminDWiki
Vitamin D: A D-Lightful Vitamin for Health – Holick Dec 2012 Korea - which has many of the same figures & tables
Evidence for vitamin D – Holick June 2012 Dermato-Endocrinology
Vitamin D books - 2 volume 2nd edition edited by Holick - Dec 2012
VITAMIN D: A D-LIGHTFUL SOLUTION FOR GOOD HEALTH – Holick 2012
Conclusions from second part are duplicated here:
Conclusions
Vitamin D exerts rapid and genomic effects in primary muscle cells and cell lines. These effects relate to intracellular calcium handling,differentiation and contractile protein composition.
In vivo, it is not clear whether VDR is expressed in adult skeletal muscle.
Whole-body VDRKO mice and vitamin D-deficient animals display significant defects in muscle function and development.
In humans, single nucleotide polymorphisms in the gene encoding VDR have been associated with differences in muscle strength.
Changes in muscle morphology in humans with severe vitamin D deficiency have been reported since the 1970's.
Proximal myopathy and muscle pain in subjects with severe vitamin D deficiency resolve following vitamin D supplementation.
Associations between vitamin D deficiency, muscle weakness and falls are confounded by factors including frailty and lower exposure to sunlight.
- Clinical parameters of muscle function are not standardized making data aggregation difficult.
Randomized data suggest that vitamin D supplementation may reduce falls in older individuals but not all studies support this conclusion.
The recommended dose of vitamin D supplementation and vitamin D targets remain hotly contested issues.
Outstanding questions
Does the VDR exist in fully differentiated adult muscle and does it have physiological relevance at this site?
- Or rather, as suggested by in vitro studies, is its role predominantly related to the function of immature muscle cells such as in myogenesis?
Are changes in muscle function and morphology directly related to vitamin D or indirectly to its effects in calcium and mineral homeostasis?
Does skeletal muscle possess the ability to 1-a-hydroxylate 25D at any stage in its development?
- As suggested by studies on phosphate handling in myocytes, does 25D itself exert direct effects on muscle?
- Is vitamin D deficiency or its reversal an important consideration among those with other muscle disorders such as congenital dystrophies and acquired immune-related myositis?
Beyond its established role in bone and mineral homeostasis, there is emerging evidence that vitamin D exerts a range of effects in skeletal muscle. Reports of profound muscle weakness and changes in the muscle morphology of adults with vitamin D deficiency have long been described. These reports have been supplemented by numerous trials assessing the impact of vitamin D on muscle strength and mass and falls in predominantly elderly and deficient populations. At a basic level, animal models have confirmed that vitamin D deficiency and congenital aberrations in the vitamin D endocrine system may result in muscle weakness. To explain these effects, some molecular mechanisms by which vitamin D impacts on muscle cell differentiation, intracellular calcium handling, and genomic activity have been elucidated. There are also suggestions that vitamin D alters muscle metabolism, specifically its sensitivity to insulin, which is a pertinent feature in the pathophysiology of insulin resistance and type 2 diabetes. We will review the range of human clinical, animal, and cell studies that address the impact of vitamin D in skeletal muscle, and discuss the controversial issues. This is a vibrant field of research and one that continues to extend thefrontiers of knowledge of vitamin D's broad functional repertoire. (Endocrine Reviews 34: 33-83, 2013)
^I. Introduction
II. Background Physiology
A. The vitamin D pathway
B. Skeletal muscle physiology
C. Calcium and muscle contraction
D. Calcium and exercise-related glucose uptake
E. Calcium and insulin-stimulated glucose uptake
III. Vitamin D and Muscle: Cell Models
A. VDR in muscle
B. Calcium homeostasis
C. Phosphate homeostasis
D. Proliferation and differentiation
E. Muscle contractile proteins
F. Phospholipid composition
G. Bone-muscle cross talk and vitamin D
H. Cell models and molecular pathways for insulin signaling and diabetes
IV. Vitamin D and Muscle: Studies in Animal Models
A. VDRKO mice
B. Other animal models
C. Animal studies on insulin sensitivity and diabetes
D. Summary: vitamin D and muscle function in animal studies
V. VDR Polymorphisms and Muscle Function
A. FokI polymorphisms
B. BsmI polymorphisms
C. VDR polymorphisms and insulin resistance/type 2 diabetes
VI. Vitamin D and Muscle: Human Studies
A. Myopathy
B. Myalgia and vitamin D deficiency
C. Fibromyalgia
D. Drug-related myopathy and vitamin D
E. Falls and vitamin D
F. Muscle strength and physical performance G. Muscle morphology and electromyography (EMG)
H. Insulin sensitivity and glucose handling
VII. Conclusions
^
Abbreviations:
AA, Arachidonic acid; AC, adenylyl cyclase; BMI, body mass index; CAMK, Ca2+/calmodulin-dependent protein kinase; CPK, creatine phosphokinase; CYP, cytochrome P450; 1,25D, 1,2 5-dihydroxyvitamin D; 25D, 25-hydroxyvitamin D; DAG, diacyl-glycerol; DBP, vitamin D-binding protein; DEXA, dual-energyx-rayabsorptiometry; EMG, electromyography; FFA, free fatty acid; FGF23, fibroblast growth factor 23; GLUT4, glucose transporter4; HbA1c, glycated hemoglobin; HOMA-IR, homeostasis model assessment of insulin resistance; IP3, inositol triphosphate; IRS, insulin receptor substrate; MHC, myosin heavy chain; MRI, magnetic resonance imaging; myf5, myogenic transcription factor 5; ODN, oligodeoxynucleotides; 1«-OHD3,1«-hydroxyvitamin D3; PKA, protein kinase A; PKC, protein kinase C; RCT, randomized controlled trial; RXR, retinoid X receptor; SNP, single-nucleotide polymorphism; SOCE, store-operated calcium entry; SR, sarcoplasmic reticulum; SRC, steroid receptor coactivator complex; STST, sit-to-stand test; TUAG, timed up and go test; VDCC, voltage-dependent calcium-channel; VDR, vitamin D receptor;
VDRE, vitamin D response element; VDRKO, VDR knockout.
I. Introduction
In recent times, there has been a great deal of interest in vitamin D, with over 1000 publications in PubMed in 2011 alone. A remarkable number of studies dealing with novel aspects of its biological activity and its potential to exert broad-ranging effects beyond calcium and mineral homeostasis have emerged (1-4). Vitamin D deficiency is a highly prevalent condition in the developed world and in the populous regions of Asia, India, and the Middle East (5, 6). Significant downward trends in vitamin D status in U.S. population-based studies suggest that vitamin D deficiency/insufficiency is increasing in frequency (7, 8). Accordingly, health agencies including the International Osteoporosis Foundation, The Endocrine Society, and Institute of Medicine have recently outlined recommendations for the prevention of vitamin D deficiency and have called for further research to guide the field (9-11).
Beyond the classic effects on bone and calcium health, the effects of vitamin D are a matter of considerable debate and have been recently reviewed in detail (12). A recent Institute of Medicine report contended that the evidence in support of an extraskeletal role for vitamin D was "not yet compelling" (11). However, there is a large and expanding body of observational data about associations between vitamin D deficiency and diverse medical conditions, ranging from multiple sclerosis to malignancy (3). Reports of the presence of the vitamin D receptor (VDR) in almost every tissue strengthen the case in favor of direct extraskeletal functions (13). The effective use of active vitamin D and vitamin D analogs in the treatment of the skin disorder, psoriasis, demonstrate that skin is an extraskeletal target tissue for vitamin D.
Long before the recognition of UV radiation as an essential component in the synthesis of vitamin D, the sun's rays were considered a source of physical strength and vitality. Ancient Egyptians revered the Sun-God, Amon-Rah, whose rays could make "a single man stronger than a crowd" (14). Herodotus recommended solaria in Ancient Greece as a cure for "weak and flabby muscles," and ancient Olympians were instructed to lie exposed and train under the sun's rays (15).
In 1952, Spellberg (16), a German sports physiologist, conducted an extensive study examining the effects of UV irradiation on elite athletes. He informed the German Olympic Committee that UV irradiation had a "convincing effect" on physical performance. This was consistent with earlier studies that reported improvements in speed and endurance among students after treatment with sunlamps (17, 18).
We have known for more than 30 yr that vitamin D exerts effects on muscle cells at a molecular level. In this journal in 1986, Ricardo Boland reviewed the effects of vitamin D on calcium handling, mineral homeostasis, and signaling pathways in muscle cells (19). Since that time, we have gained further insight in its effects on the regulation of cell survival (20), differentiation (21), and calcium handling (22). In more recent times, clinical studies have examined the effects of vitamin D supplementation on muscle function and falls in various populations (23, 24).
However, the field is challenged by controversy. A recent report suggesting that VDR was not detectable in muscle has fueled the debate as to whether vitamin D effects on muscle are direct or indirect (25-27). The creation of the VDR knockout (VDRKO) mouse in 1997 gave a new focus to this question, which we will discuss (28). A continuing area of uncertainty stems from conflicting meta-analyses of clinical studies examining the effect of vitamin D supplementation on muscle strength and falls in older individuals (23, 29-31).
In this review, molecular, animal, and human studies examining the various roles of vitamin D in muscle will be presented. We will discuss contentious issues that have made this a vibrant field of research and one that continues to extend the frontiers of our knowledge of vitamin D's broad functional repertoire.
II. Background Physiology
A. The vitamin D pathway
The family of molecules known collectively as vitamin D are not true vitamins, which are defined as essential substances obtained exclusively from the diet. The misnomer is a remnant of the early work of a number of scientific pioneers from the 1900s.
After inducing rickets in a group of dogs by keeping them indoors for prolonged periods, the British physician Sir Edward Mellanby (32) discovered that feeding them cod-liver oil cured them and attributed this to the recently identified vitamin A (33). However, in 1922, McCollum et al. (34) showed that after heating and aerating cod-liver oil to destroy the vitamin A, it remained effective in the treatment of rickets but no longer cured night blindness. Mc-Collum followed the sequential alphabetical designations and labeled the new substance "vitamineD." In the 1920s, it was recognized that children with
Figure 1.
Summary of the molecular structures and various names of vitamin D and its related compounds (molecular structures taken from PubChem Public Chemical Database www.ncbi.nlm.nih.gov/pccompound).
"rapid disappearance" of their illness and improved "general vigor and nutrition." This finding was the direct extension of earlier work by Huldschinsky (36), who achieved the same outcome with artificially produced UV Light.
After these seminal studies, Harry Goldblatt and Kath-erine Soames (37) reported in 1923 that the irradiation of certain foodstuffs rendered them antirachitic. In 1926, Adolf Windaus etal. (38) identified the chemical structure of cholecalciferol (vitamin D3) as found in irradiated pig skin as well as the structure of its parent molecule, 7-dehydrocholesterol. Windaus also isolated vitamins D1 and D2 and was awarded the 1928 Nobel prize for his work on sterols and vitamins. The nomenclature in the field is often confusing. Names, alternate names, and molecular structures of vitamin D and related molecules are shown in Fig. 1. The metabolic pathway of vitamin D, including the various steps in its activation and degradation, are depicted in Fig. 2.
Figure 2.
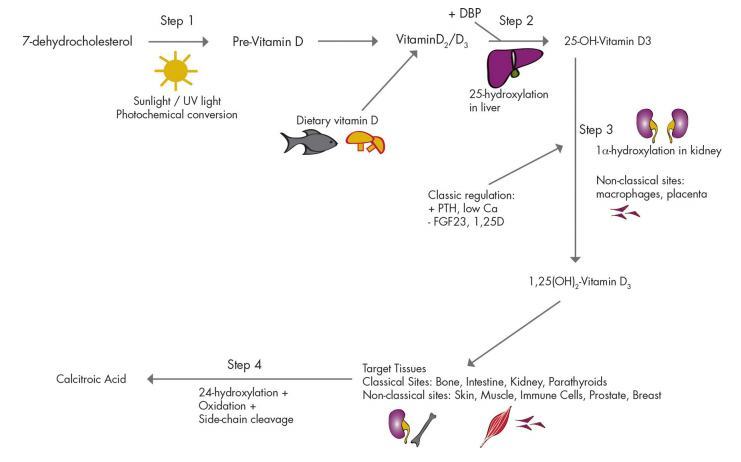
Figure 2. Summary of the vitamin D pathway. Steps are discussed in the text. Molecular structures and alternate names for each of the compounds are found in Fig. 1.
It was assumed that exposure of the skin to UV radiation drove the conversion of 7-dehydrocholesterol to cholecalciferol (step 1, Fig. 2). However, proof of this emerged more than 30 yr later with independent discoveries by two groups: Holick etal. (39) andEsvelt etal. (40). The photo-production is subject to a variety of factors including amount of UV exposure (latitude, season, and use of sunscreen and clothing), ethnicity (skin pigmentation), and age (41-43). After the photochemical conversion of 7-dehydrocholesterol to pre-vitamin D3 and its thermal isomerization to vitamin D3, it binds to the vitamin D-binding protein (DBP), and is transported to the liver where a hydroxyl group is attached at the carbon-25 atom (i.e. C-25) to generate 25-hydroxyvitamin D (25D) (step 2, Fig. 2). The importance of the liver in this first phase of hydroxylation was reported in 1969 by DeLuca and colleagues (44). A number of 25-hydroxylases have been reported including cytochrome P450 CYP27A1 and CYP2R1 (45,46). CYP2R1 is probably the major enzyme required for 25-hydroxylation of vitamin D3, at least in humans (47, 48). A patient with classic rickets and low circulating levels of 25D was found to have a homozygous mutation of the CYP2R1 gene, implying that other enzymes were unable to compensate (49).
Much remains unknown about the 25-hydroxylase enzymes including the significance of their reported presence in skin, kidney, and intestine (50). These enzymes are generally considered to be constitutively expressed with little feedback regulation; however, this is unusual for the CYP family (50). In the absence of severe hepatic dysfunction, 25-hydroxylation of vitamin D is not usually rate limiting. However, in mild to moderate liver impairment, the associated fat malabsorption can cause vitamin D deficiency.
In contrast, 1 a-hydroxylation is the major rate-limiting step in synthesis of 1,25-dihydroxyvitamin D (1,25D) (step 3, Fig. 2). Synthesis of 1,25D is tightly regulated (51) and is mediated by the enzyme 1a-hydroxylase. Factors regulating 1a-hydroxylase in kidney are shown in Fig. 2. Due to its sequence similarity to CYP27A1, the gene encoding 1a-hydroxylase was called CYP27B1 (52). Its role was demonstrated in 1998 by the development of rickets and reduced circulating 1,25D levels in four patients with gene mutations for this enzyme (53). Cyp27B1-null mice also develop rickets with reduced levels of circulating 1,25D (54).
CYP27B1 mRNA is expressed in a number of vitamin D target tissues including kidney, skin, intestine, macro-phages, and bone. Although its expression is relatively high in skin, the kidney is thought to be primarily responsible for circulating levels of 1,25D (50). This is supported by 1,25D deficiency in people with renal failure (55, 56). However, this has not been conclusively proven with renal-specific CYP27B1 deletion. The presence of CYP27B1 in other cell types, especially macrophages, is demonstrated by the fact that people with granulomatous diseases can have elevated 1,25D levels (57).
Entry of 25D into the proximal renal tubular cells requires receptor-mediated uptake of DBP plus 25D at the brush border, degradation of DBP by legumain, and en-docytic internalization and translocation of 25D to mitochondria (58). Megalin, a member of the low-density li-poprotein receptor family, is required for reabsorption of filtered DBP-bound 25D (59). It is in the mitochondria that 1 a-hydroxylation of 25D into its biologically active form, 1,25D, occurs (50). A number of factors contribute to the tight regulation of 1a-hydroxylase enzyme expression and activity in the kidney (Fig. 2). These include calcium, PTH, calcitonin, GH, IGF-I, and fibroblast growth factor23 (FGF23).In addition, 1,25D negatively regulates its own synthesis by suppressing 1a-hydroxylase expression in kidney and bone (60). There is also evidence to suggest that estrogen, progesterone, and prolactin may regulate 1a-hydroxylase activity (61, 62). In macrophages, regulation of CYP27B1 is primarily cytokine mediated (63).
The final important enzyme in the vitamin D endocrine system is 24-hydroxylase (CYP24A1). Found in nearly all cells and highly expressed by the kidney, CYP24A1 limits the amount of 1,25D in target tissues by converting 1,25D to inactive metabolites, including 1,24,25-(OH)3D and calcitroic acid and by converting 25D to 24,25(OH)2D (step 4, Fig. 2). In addition to 24-hydroxylation, this multicatalytic enzyme is able to catalyze side-chain hydroxy-lations at the C23 and C26 positions (64). Recently, mutations in CYP24A1 were reported in six children with infantile hypercalcemia, thereby providing conclusive evidence of the importance of this enzyme in the in vivo regulation of vitamin D metabolism (65).
Figure 3.
Figure 3. Classic vitamin D signaling pathway. Vitamin D binds to its receptor, which dimerizes, preferentially with RXR. This complex binds to VDRE in the DNA to regulate transcription.
The VDR, to which 1,25D binds to exert its biological effects, was described in 1974 by Brumbaugh and Haussler (66). This is depicted in Fig. 3. Insights into the structure and function of this protein have been gained via the cloning and subsequent analysis of the recombinant protein (67), by x-ray crystallography (68) and molecular modeling using atomic coordinates of the protein x-ray structure (69). The protein comprises three distinct regions: an N-terminal dual zinc finger domain that binds to DNA (a characteristic feature of the steroid receptor family), a C-terminal domain that binds to 1,25D, and an extensive, unstructured region that links these two functional domains.
Binding of 1,25D to VDR leads to conformational changes within the receptor that allows it to interact with its heterodimeric partner, retinoid X receptor (RXR) (Fig. 3) (70). VDR also forms homodimers that bind DNA and regulate gene expression (71). The liganded complex (i.e. 1,25D-VDR-RXR) binds to vitamin D response elements (VDRE) in the DNA (72). Classic VDRE are direct repeats of two hexameric core binding sites with a three-nucleotide separation (73, 74). However, numerous nonclassic sites have been proven to act as VDRE.
VDR-containing dimers interact with large coregulatory complexes required for gene modulation (70, 75). Although a number of coregulatory molecules have been characterized including the VDR interacting protein and the steroid receptor coactivator complex (SRC), the precise mechanisms by which these molecules operate are just beginning to emerge (76). The system is more complex, because VDR is one of the few nuclear hormone receptors that has been clearly demonstrated to be able to regulate gene expression in the absence of ligand. Unliganded VDR dimers can bind to and regulate some genes, mostly to repress their expression. This is thought to be the mechanism for spontaneous hair loss in mice with mutations in VDR (77). Expression of the VDR in virtually every tissue and the diverse phenotypic changes in the VDRKO mouse are consistent with the wide spectrum of activity of the 1,25D-VDR endocrine system (13).
As well as regulation of VDR, CYP27B1, and CYP24A1 (78, 79), the 1,25D-VDR-RXR complex is involved in regulation of a variety of cellular functions including DNA repair, cell differentiation, apoptosis, metabolism, and oxi-dative stress (13). Its effects on calcium and mineral homeostasis are well established and, in brief, result from the transcriptional regulation of specific proteins within the intestine (calcium-binding proteins, calbindin D28k, and epithelial calcium channels), bone (osteocalcin, osteopontin, and receptor activator of nuclear factor-KB ligand), and parathyroid glands (PTH) (80). These effects provide potential indirect routes for regulation of muscle function in addition to direct effects (Fig. 4).
This review will focus on diverse effects of the vitamin D endocrine system on the functional and metabolic capacity of skeletal muscle as reported by a range of clinical and translational studies. We will also discuss the central role of skeletal muscle in our emerging understanding of the nongenomic capabilities of the VDR.
B. Skeletal muscle physiology
Skeletal muscle is estimated to account for 42% of total body mass in males and 35% in females (81). Its primary function is to generate force and to provide locomotion. The functional units of skeletal muscle are the muscle fibers, themselves comprised of many myofibrils. Myofibrils are long cylindrical multinucleated cells that vary considerably in their morphological, biochemical, and physiological properties, thereby forming the basis of the well-known structural and functional diversity of skeletal muscle (82). This complexity causes difficulty in classifying muscle fibers. At one stage, one author described fiber classification as "showing an alarming trend toward the incomprehensible" (83).
At this time, the most widely used classification is based on histochemical methods that determine the pH lability of myofibrillar ATPase activity and divides fibers into type I (low activity) and type II (high activity) with further subdivision into IIA, IIX, and IIB (84) depending on the expression of different myosin heavy chain (MHC) forms (summarized in Table 1). The reliance on oxidative or glycolytic metabolic pathways determines the contractile speed of these various fiber types. There is substantial evidence that muscle fibers are dynamic in their response to a variety of contractile and metabolic stimuli and are able to convert from one fiber type to another or undergo atrophy (84). Both vitamin D deficiency and age-related sarcopenia have been associated with preferential atrophy of type II (fast-twitch) fibers (84, 85).
On a macroscopic scale, the generation of force by a muscle is dependent on several factors including size, fiber composition, and individual fiber functional capacity. The cross-sectional area is the sum of the individual, parallel fibers, themselves made up of thousands of individual myofibrils and other cell types (86). The sarcomere is the basic unit of contraction and is defined as the portion of the myofibril that lies between two bands, known as Z bands. Between successive Z bands, an array of myosin and actin molecules are intricately arranged to form alternating filaments, suspended in the sarcoplasm and lying in close proximity to mitochondria, indicative of the significance of ATP in contraction. The role of calcium, also fundamentally important in the tight regulation of muscle contraction (87), will be discussed below.
Apart from the generation of force, skeletal muscle is a highly metabolic tissue that produces and responds to a variety of hormones and factors, leading one author to describe it as a true endocrine organ (88).
Figure 4.
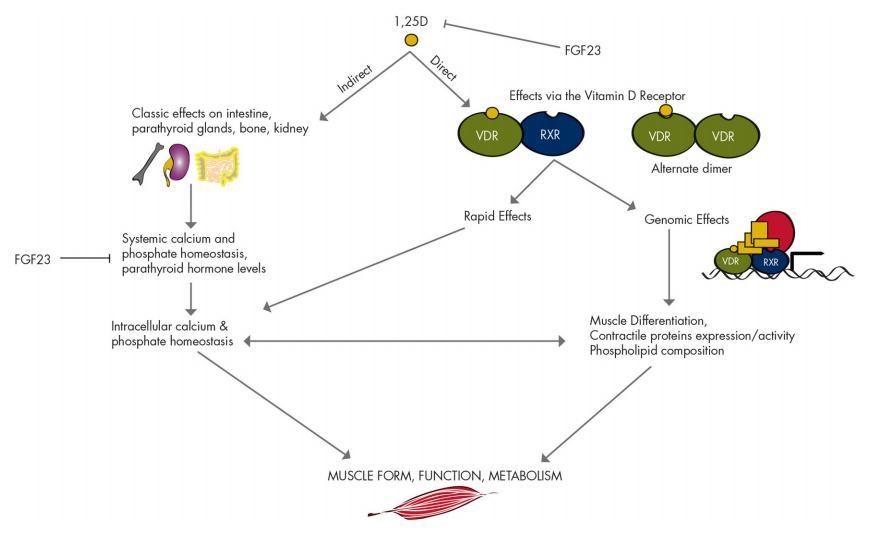
Figure 4. Direct and indirect effects of vitamin D on muscle. Data on direct effects come predominantly from in vitro studies and are yet to be confirmed in vivo, where the presence of the VDR is currently under debate.
Exercise leads to the increased expression and secretion of a family of myokines including IL-6 and brain-derived neurotrophic factor that can stimulate glucose uptake and fat oxidation within muscle, lipolysis in adipocytes, and gluconeogenesis in hepatocytes via various autocrine, paracrine, and endocrine pathways (87, 89). Skeletal muscle is also responsive to a range of hormones including, but not limited to, insulin, IGF, glucocorticoids, thyroid hormones, and 1,25D, all of which exert influences on the differentiation, metabolism, and function of muscle via a number of established and evolving mechanisms.
C. Calcium and muscle contraction
As well as regulating whole-body calcium homeostasis, there is also evidence that 1,25D increases calcium influx in muscle cells and thus may have both direct and indirect calcium-related effects on muscle (Fig. 4) (90). The sliding filament theory, first proposed in 1954 (91), describes the highly complex movement of actin and myosin filaments over each other. This and potential effects by which vitamin D may affect this model, based on data to be discussed, are depicted in Fig. 5. It is primarily the influx of calcium from the sarcoplasmic reticulum (SR) and binding to the troponin-tropomyosin complex that results in the exposure of active binding sites on the actin filament and their engagement with the myosin heads (82). In the presence of ATP, contraction ensues as the myosin head tilts from an obtuse angle to the perpendicular, causing movement of myosin over actin filaments in a process named the power stroke, after which ADP and inorganic phosphate are released (86). The binding of new ATP to the myosin head causes its detachment from the active site of the actin filament and movement back into the obtuse position.
TABLE 1. Respective characteristics of muscle fiber types based on the current classification
Figure 5.
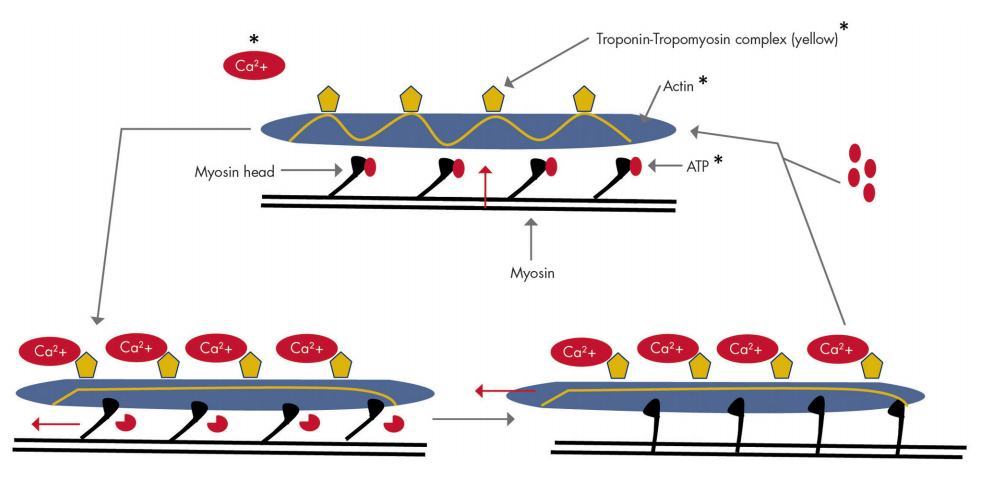
Figure 5. Potential roles of vitamin D in the sliding-filament model of muscle contraction. These potential effects, marked with an asterisk, include the rapid flux of calcium ions from the SR to cytoplasm (319), the expression of actin and troponin-tropomyosin proteins (1 52, 1 53), and the effects of vitamin D-mediated phosphate homeostasis on ATP (326).
D. Calcium and exercise-related glucose uptake
Calcium plays a vital role in exercise-related glucose uptake by skeletal muscle. Vitamin D regulates calcium homeo-stasis, giving potential for indirect regulation. Exercise increases glucose transporter 4 (GLUT4) expression; after contraction, increased cytosolic calcium activates Ca2+/cal-modulin-dependent protein kinase (CAMK) pathways and causes transcriptional up-regulation of myocyte enhancer factors 2A and 2D, which increase GLUT4 expression (9294). Exercise also increases GLUT4 translocation to the muscle cell membrane, independently of insulin. Activation of the AMP-kinase pathway contributes to this process (95). GLUT4 vesicle translocation and insertion into the cell membrane is a calcium- and ATP-dependent process (96). A putative mechanism for this is the synaptotagmins. They are calcium-sensitive proteins required for insertion of the GLUT4 proteins into the cell membrane, as demonstrated in adipocytes (97). Synaptotagmins in turn regulate Myo1c, an actin-filament-attached protein that binds to and transports GLUT4 vesicles (98). Contraction-induced calcium influx stimulates a range of signaling pathways that regulate muscle differentiation and function (99, 100). These include myogenic transcription factors, myostatin, peroxisome proliferator-activated receptor 8 and utrophin A, mainly via CAMK and calcineurin-mediated pathways (82, 99, 101). These processes, generally referred to as excitation-transcription coupling, give rise to the plasticity and unique adaptive ability of muscle to alter vital components in its function, fiber type, and contractile force on demand.
E. Calcium and insulin-stimulated glucose uptake
Skeletal muscle is responsible for approximately 85% of insulin-mediated glucose uptake in lean individuals (102). Insulin induces translocation of GLUT4 to the cell surface, facilitating glucose uptake and clearance of circulating glucose. Insulin binds to the a-subunits of its receptor, activating a signaling cascade. This has been covered in many elegant reviews (103,104). The mechanisms by which the activation of these proteins then leads to the insertion of GLUT4 protein into the cell membrane remain incompletely understood but have also been the subject of recent review (105). As with exercise, GLUT4 vesicle translocation is ATP and calcium dependent.
Pharmacological inhibition of calcium influx in muscle reduces insulin-mediated glucose uptake, independent of effects on Akt (96). Calcium regulates components of the proximal insulin signaling pathway such as the binding of calmodulin to insulin receptor substrate (IRS)-2 (106). Increases in calcium influx improved insulin-mediated glucose uptake in isolated muscle fibers of both normal and insulin-resistant mice (96). Calcium regulates cytoskeletal components involved in GLUT4 translocation (98). Studies on L6 myotubes reported significant increases in GLUT4 expression in response to caffeine-related increases in intracellular calcium. The effects were negated by dantrolene, an inhibitor of SR calcium release (92). However, additional research examining the role of calcium in insulin sensitivity is needed.
III. Vitamin D and Muscle: Cell Models
On a cellular level, a variety of mechanisms by which vitamin D impacts upon the function of skeletal muscle have been elucidated. These can be broadly divided into 1) genomic effects that arise from the binding of the 1,25D-VDR-RXR heterodimer at specific nuclear receptors to influence gene transcription and 2) nongenomic effects that arise from a host of complex intracellular signal transduction pathways after binding of 1,25D to nonnuclear receptor. Over the past 30 yr, the majority of research in this area has mainly focused on the nongenomic effects of vitamin D on skeletal muscle, in particular its regulation of protein kinase A (PKA)/cAMP, protein kinase B, protein kinase C (PKC), CAMK, and multiple MAPK pathways (90).
A. VDR in muscle
After the discovery of the VDR in 1969 (107), the isolation of unoccupied 1,25D receptors partitioned between the cytosol and cell nucleus in intestinal cells in1980raised the possibility of rapid, nontranscriptional pathways associated with this receptor (108). The rapidity, over minutes, with which 1,25D treatment resulted in changes in intracellular calcium transport in vascularized duodenal cells supported this possibility (109). Furthermore, it became apparent from studies that examined the binding properties of VDR isolated from the caveolae-enriched membrane fraction of chick intestinal cells that the cytosolic receptors were identical to nuclear VDR (110). To confirm this, significant reductions in the capacity of [3H]1,25D to bind to isolated caveolae-membrane fractions were reported in tissues obtained from VDRKO mice (110). Studies examining the Tokyo strain of VDRKO mice, in which the second zinc finger of the DNA-binding domain of VDR is ablated, reported some residual binding of 1,25D in kidney cells (111).
Treatment with 1,25D elicits rapid uptake of calcium within muscle cells in vitro and in vivo (112). However, after transfection of muscle cells with anti-VDR antisense oligodeoxynucleotides (ODN), 1,25D-dependent mechanisms by which rapid calcium entry occurs, namely store-operated calcium entry (SOCE) or capacitative calcium entry, are inhibited (113,114), therefore implying a direct nongenomic role for VDR in calcium handling. The presence of the VDR has been reported in avian, murine, and human muscle cells on the basis of immuno-histochemistry (20, 115), equilibrium binding studies (25), and detection of VDR mRNA by RT-PCR (21).
However, these findings are subject to challenge due to the nonspecificity of many of the VDR antibodies (116). A recent paper that used a validated antibody (which did not show bands in VDRKO mice) did not find VDR expression in skeletal, cardiac, or smooth muscle by Western blot and immunohistochemistry (27). Differences in experimental conditions and the possibility of tight protein binding of VDR to DNA may have accounted for this finding. Moreover, low levels may be sufficient for significant function in muscle. Major studies examining the presence of VDR in muscle and the various techniques used have been summarized in Table 2.
Another possibility is that there may be differences in the expression of VDR in muscle in different species and throughout the various stages of muscle differentiation. In support of the latter, in vitro studies predominantly examine the activity of VDR within myoblasts and myotubes rather than fully differentiated adult cells. In vivo, VDR mRNA was reported in the muscle of 3-wk-old wild-type mice but not in their 8-wk-old counterparts (21). The authors suggested a primary role for VDR in early muscle development (21). Thus, the expression of VDR within muscle over time requires further clarification.
Within cultured chick myoblasts, VDR translocates from the nucleus to the cytoplasm quite rapidly (i.e. 1-10 min) after exposure to 1,25D (117). Intact microtubular transport and caveolae structure were essential as demonstrated by the disruption of VDR translocation by inhibition of these individual components in C2C12 cells (i.e. with colchicine and methyl-j3-cyclodextrin, respectively) (20). After a longer period of exposure to 1,25D, the VDR appears to translocate back to the nucleus to presumably carry out its role in transcriptional regulation. This shuttling of the VDR between cytoplasm and nucleus indicates its versatility in both rapid and genomic actions, depending on location (118).
Coimmunoprecipitation analyses have also demonstrated direct binding of the VDR with a component of the tyrosine phosphorylation cascade, namely c-Src, under the influence of 1,25D (119). Transfection of muscle cells with three different anti-VDR antisense ODN inhibited 1,25D-dependent dephosphorylation and subsequent activation of c-Src as assessed 15 min after treatment with 1,25D or vehicle (120). More recently, treatment of muscle cells with 1,25D resulted in VDR binding with c-Src, time-dependent increases in c-Src activity, and the redistribution of c-Src from the periplasma membrane zone, where it resides under basal conditions, to the cytoplasm and nucleus as seen on confocal microscopy (20). There is also evidence that 1,25D induces the association between c-Src and c-myc, a transcription factor involved in cell growth and apoptosis. This evidence includes coimmunoprecipi-tation analyses that demonstrate 1,25D-mediated formation of complexes between c-Src and c-myc (20) and significant inhibition (i.e. 94%) of 1,25D-mediated c-myc tyrosine phosphorylation on immunoblot after transfec-tion of muscle cells with anti-VDR antisense ODN (120) or treatment with PP1, a c-Src-specific inhibitor (120). Mechanistically, c-Src probably interacts with VDR and c-myc, both of which are hormone-dependent phospho-tyrosine proteins, via its Src homology 2 domain, but this requires further evaluation (121). A role for caveolin-1 in 1,25D-mediated activation of c-Src has also been suggested. Caveolin-1 belongs to a family of membrane-scaffolding proteins with potential roles in different disease phenotypes and binds to c-Src under basal conditions near the cell membrane. After treatment of C2C12 myoblasts with 1,25D, colocalization of caveolin-1 and c-Src was disturbed, and they were redistributed into cytoplasm and nucleus (20). Interestingly, when the caveolae structure was disrupted by methyl-j3-cyclodextrin, 1,25D was not able to separate caveolin-1 from c-Src, preventing its activation and also preventing the nuclear translocation of VDR. Therefore, the caveolae and associated proteins appear to play an upstream role in the activation of c-Src. It also appears that 1,25D-mediated activation of c-Src by VDR is a downstream regulator of several nongenomic effects of 1,25D in muscle, specifically involving differentiation and calcium homeostasis, which we will discuss.
Apart from the specific ability of the VDR to translocate from nucleus to cytosol in response to 1,25D, conforma-tional changes within the highly flexible 1,25D molecule also determine the mediation of genomic and nongenomic actions via its receptor. It has been shown that the relatively planar 6-s-cis locked JN [1,25-(OH)2-lumisterol3] displays nongenomic activity and that the 6-s-trans locked JB [1, 25-(OH)2-dihydrotachysterol3] possesses predominantly genomic activity on binding the VDR (69). By molecular modeling of the VDR using atomic coordinates of the protein x-ray structure and computer docking, specific binding sites on the VDR that determine its functional activity have been reported (122, 123). Interestingly, the pockets that, respectively, mediate the regulation of nongenomic and genomic responses overlap and therefore result in mutually exclusive conformational forms of the VDR. It has been suggested that the unbound VDR may possibly exist in the cytoplasm as multiple, equilibrating receptor conformations according to standard statistical distribution (69). Also intriguing, a potential role for 25D in binding the alternative pocket and initiating intracellular calcium flux has recently been reported (123).
Apart from the VDR, it is also possible that other cytosol receptors may be responsible for the rapid actions of 1,25D in muscle. Contrary to an earlier report, it does not appear that annexin II binds to 1,25D in a physiologically relevant manner (124,125). Recent data have suggested a role for membrane-associated rapid response steroid binding in potentially inducing rapid effects of 1,25D in muscle, but further assessment is required (126).
B. Calcium homeostasis
Although the nongenomic regulation of intracellular calcium by 1,25D has been well characterized in cultured myoblasts and myotubes, it has also been confirmed by in vivo studies on chicks and in vitro assessment of differentiated soleus muscle samples. Studies report a time- and dose-dependent increase in intracellular muscle calcium uptake in response to 1,25D. The use of particular agents and antisense oligonucleotides to block components of the signal transduction pathway has led to the elucidation of specific step-wise mechanisms by which vitamin D influences intracellular calcium homeostasis (127-129). These have been depicted in Fig. 6.
First, the rapid mobilization of calcium from the SR into the cytosol relies on 1,25D-dependent activation of two components of the signal transduction pathway, namely c-Src and phosphoinositide-3 kinase. These in turn lead to the activation of phospholipase Cy, the rapid production of inositol triphosphate (IP3) and the first of two phases of diacylglycerol (DAG) synthesis from the membrane phosphoinositides (22, 130). It is IP3 that then mediates the rapid movement of calcium from the SR into the cytosol (130).
In the continued presence of 1,25D, two additional processes are then involved in the more sustained phase of calcium entry from the extracellular compartment, namely SOCE and L-type voltage-dependent calcium-channel (VDCC) entry mechanisms. SOCE relies on several factors including IP3-dependent calcium release from the SR that activates calmodulin, CAMKII, and PKC, the latter also being activated by a by-product of a previous reaction, namely DAG (131). The particular channels responsible for SOCE have been identified as the transient receptor potential-canonical-like proteins. Interestingly, a direct role of the VDR in activating these channels has been suggested by the coimmunoprecipitation of both molecules after treatment with 1,25D in chick skeletal muscle cells (113).
Figure 6.
Figure 6. Mechanisms by which vitamin D influences calcium homeostasis in cultured muscle cells. c-Src, cellular Src; PI3-kinase, phosphoinositide 3-kinase; PKA, protein kinase A; PLC-y, phospholipase Cy.
Apart from its reported role in SOCE, evidence supports vital additional roles for PKC in 1,25D-dependent calcium homeostasis in muscle. Rapid translocation of PKC-a from the cytosol to the cell membrane after the in vitro treatment of chick soleus muscle and cultured rat and chick myoblasts with 1,25D (132, 133) and marked reduction in intracellular calcium influx after selective knockout of PKC-a by the use of specific antisense oligo-nucleotides have been described (128). Furthermore, PKC also activates VDCC-mediated calcium entry as evidenced by the rapid stimulation of 45Ca2+ uptake by cultured myoblasts after treatment with PKC activators, namely DAG and phorbol 12-myristate 13-acetate, inhibited by the addition of nifedipine, an L-type VDCC blocker (133). PKC-a may also have a role in the 1,25D-dependent activation of the ERK1/2 signaling pathway as will be discussed in Section III.D on proliferation and differentiation.
VDCC-mediated calcium entry also relies on 1,25D-dependent activation of the cAMP/PKA pathway. Very rapid increases (within 30 sec) in the levels of adenylyl cyclase (AC) and cAMP levels, together with increased PKA activity, occur in differentiated muscle cells and cultured myotubes after 1,25D treatment (127, 134). These studies also report that 1,25D stimulation of VDCC-45Ca2 + entry can be mimicked by treatment with dibutyryl cAMP and abolished by specific inhibitors of AC and PKA.
Another mechanism is emerging by which PKC and cAMP/PKA pathways may cross talk in the regulation of VDCC-mediated calcium flux (90). The preliminary data indicates an increase in the cAMP content of myoblasts after treatment with a PKC activator, phorbol 12-myristate 13-acetate, which may stem from the phosphorylation of Gai that is mediated by PKC in other cells (133). Furthermore, the phosphorylation of Gai in myoblast membranes after treatment with 1,25D is likely to be essential in the stimulation of AC activity as evidenced by the effects of abolishing G protein regulatory pathways on 1,25D-mediated AC activity (134).
In summary, 1,25D induces changes in intracellular calcium levels in cultured muscle cells initially via rapid IP3-dependent calcium shifts from the SR to the cytosol followed by processes resulting in extracellular calcium influx via the activation of SOCE and VDCC activity. These mechanisms are evident in both immature myoblasts and differentiated myotubes, suggesting a potential role for rapid calcium influx in muscle cell differentiation and contraction, respectively (19). In support of this inference, an interesting report from 1974 described a direct correlation between in vivo skeletal muscle dysfunction and demonstrable defects in intracellular calcium handling (136). A group of rabbits, rendered vitamin D deficient by dietary methods, were found to be substantially weaker and hypo-tonic compared with their vitamin D- replete counterparts and in vitro, displayed significant reductions in calcium uptake in the SR on isolation from psoas muscle.
C. Phosphate homeostasis
Phosphate is an essential substrate in the production of ATP and in protein synthesis. There is early evidence demonstrating that phosphate uptake in muscle may be influenced by 25D (19, 137). The administration of 25D to vitamin D-deficient, phosphate-deplete rats resulted in a significant increase in the in vitro concentration of [32P]phosphate in muscle cells, followed by the stimulation of phosphate-dependent metabolic processes including ATP synthesis in these cells (137). This effect was not reproduced by the repletion of phosphate in these rats. The abolition of 1a-hydroxylase activity by nephrectomy had no effect on 25D-mediated phosphate uptake, implying a direct role of the prehormone in this process. Another study reported the specificity of 25D on in vitro phosphate uptake of differentiated muscle cells with the absence of such effects after 1,25D and 24,25-(OH)2D treatment (138-140).
Another study demonstrated the specific transport of [32P]phosphate across muscle plasma membranes after in vivo vitamin D repletion and 32P labeling of vitamin D-deficient chicks (141). This finding was later confirmed by an increase in vesicle phosphate transport in muscle cells via the isolation of highly purified sarcolemma vesicles in vitamin D-deficient chicks treated with vitamin D (19). The direct effect on phosphate uptake may be mediated via a sodium-dependent mechanism as reported by studies on cultured muscle cells (138, 139).
D. Proliferation and differentiation
There is evidence that 1,25D activates components of the MAPK family in cultured myoblasts, thereby influencing the expression of genes involved in cellular proliferation and differentiation. The majority of research in this area has focused on the effects of 1,25D on the ERK1/2 signaling pathway. The initial activation of c-Src by 1,25D, previously described as an apparent gateway to the nongenomic effects of 1,25D in muscle, results in the rapid activation of Raf-1 by the phosphorylation of its serine residue, which relies on the involvement of Ras and PKC-a (142, 143).
Raf-1 then leads to the activation of MAPK kinase, which activates ERK1/2, after which the phosphorylation of a range of proteins and transcription factors including cAMP response element-binding protein and Elk-1 and the increased expression of other proteins relevant to cell proliferation and differentiation, namely c-myc and c-fos, take place (144, 145).
Another MAPK activatedby1,25D incultured myoblasts is p38. Rapid c-Src-dependent stimulation of MAPK kinases MKK3 and 6 and p38 was demonstrated in C2C12 myoblasts after treatment with 1,25D (146). After this, p38-de-pendent activation of MAPK2 and subsequent phosphory-lation of heat-shock protein 27 was demonstrated. Heat-shock protein 27 has an important role through its association with the actin microfilament system and cyto-skeletal remodeling of muscle cells (146, 147). More recently, 1,25D-mediated AKT phosphorylation in differentiating C2C12 cells was also shown to occur via c-Src, p38, MAPK and phosphoinositide-3 kinase pathways (148).
Although little is known about its exact activity in this context, a third member of the MAPK family, namely c-Jun N-terminal kinase-1/2, is also phosphorylatively activated by 1,25Din C2C12 myoblasts (146). Therefore, an intricate system of nongenomic regulatory responses to 1,25D may control cellular proliferation and differentiation of muscle cells, although the relative role of each component remains unclear.
A variety of genomic responses to 1,25D have also recently been elucidated. Apart from being the first to describe the presence of VDR within muscle cells, Simpson et al. (25) also demonstrated dose-dependent reduction in the proliferation of G-8 myoblast cells that were treated with 1,25D and a commensurate reduction in DNA synthesis, suggesting that genomic responses to 1,25D gave rise to the down-regulation in myoblast proliferation and enhanced differentiation into myotubes.
In a recent study, treatment of C2C12 myoblasts with 1,25D for 7 d as opposed to vehicle resulted in increased mRNA and protein expression of transcription factors known to enhance myogenesis, namely myogenic differentiation antigen, desmin, myogenin, and IGF-II and the reduced expression of proliferating cell nuclear antigen and myostatin, which, respectively, enhance cell proliferation and negatively regulate muscle mass (118). Morphologically, cells treatedwith1,25D for10 d displayed significantly increased muscle fiber size and diameter, as indicated by staining for MHC type II a late myogenic marker. These changes were associated with VDR-induced genomic mechanisms as evidenced by significant increases in the expression of the receptor by1dof1,25D treatment and its nuclear translocation at4das opposed to its persistent location in the cytoplasm of cells treated with vehicle.
However, these findings stand in contrast to an earlier report that described the down-regulation of myogenin and myogenic transcription factor 5 (myf5) at an mRNA level in C2C12 myoblasts treated with 1,25D for 48 and 96 h compared with those treated with vehicle (21). Furthermore, this coincided with the reduced expression of neonatal forms of MHC, suggesting myoblast maturation in cells treated with 1,25D. It is possible that differences in study design may have accounted for these contradictory findings, specifically pertaining to myogenin expression. Different durations of treatment, and daily vs. single treatment regimens were employed in these studies (21, 118).
In another study Artaza and Norris (390), vitamin D treatment of mesenchymal stem cells resulted in increased expression of follistatin, an antagonist of myostatin, and caused down-regulation of TGF-/3. These vitamin D-me-diated changes in gene expression imply that it has a potential role in the inhibition of fibrosis and, perhaps, the promotion of myogenesis and osteogenesis in mesenchymal stem cells.
In a recent study, European sea bass treated with various doses of dietary cholecalciferol from 9 -44 d after hatching demonstrated dose-dependent effects in the gene expression of a number of myogenic transcription factors and dose-dependent increases in white muscle fiber size and number (149).
Although vitamin D has clear effects on muscle differentiation, more research is needed to elucidate the nature of these mechanisms. It is also important to note that the overlapping functions and complex regulatory pathways determining the activity of myogenic transcription factors are themselves something of a mystery although recently reviewed in detail (150).
E. Muscle contractile proteins
There is evidence that vitamin D may play a role in the regulation of key components in the cytoskeletal structure of muscle cells. As discussed in Section II.B, the complex interaction between actin and myosin, two cytoskeletal proteins, forms the basis of understanding muscle contraction (Fig. 5). Similar proteins play a potential role in intracellular trafficking and, potentially, GLUT4 translocation.
Significant reductions in components of the actomyo-sin-troponin complex in the skeletal muscle of vitamin D-deficient rats and rabbits have been reported in two separate studies, although in the latter, this may not have been directly related to 1,25D because in vivo administration of ethane-1-hydroxy-1,1-diphosphate in doses known to inhibit 1,25D had no effect on these components of the cytoskeleton (151, 152). A study also reported an increase in the muscle concentrations of actin and troponin C in chicks replete with vitamin D as opposed to their vitamin D-deficient counterparts (141). Taken together, these reports suggest a direct role for 25D in the up-regulation of these muscle contractile proteins (141, 151). Furthermore, the direct role of 25D in phosphate homeostasis, as discussed, and the recent demonstration of direct binding between 25D and the alternative pocket of the VDR with subsequent biological effects in COS-1 kidney cells suggest that reconsideration of the ability of 25D to generate biological responses in vivo may be in order (123).
F. Phospholipid composition
Phospholipids, a class of lipids that reside within the cell membrane, have been implicated in a variety of signal transduction pathways, including insulin signaling and calcium handling, and play a role in cell membrane function including caveolae. Alterations in phospholipid composition have been associated with insulin-resistant states (153). There is also evidence of a direct role of 1,25D in the regulation of the phospholipid metabolic pathway.
A study reported higher relative concentrations of phospholipids in the muscle SR membranes of vitamin D-replete vs. -deficient animals (141). There were also significant changes in the levels of particular phospholipids, specifically an increased concentration of phosphatidylcholine and decreases in phosphatidylethanolamine, in the sarcolemma vesicles of vitamin D-replete vs. -deficient chicks (141). Another study suggested that 1,25D treatment led to the activation of specific methylation pathways that leads to the conversion of phosphatidylethanolamine to phosphatidyl-choline in muscle cells (154). This is likely to represent a genomic effect as 1,25D-mediated binding of [3H]glycerol and [14C]ethanolamine to phosphatidylcholine in cultured myoblasts was inhibited by the action of actinomycin D, an inhibitor of DNA synthesis. Although the precise significance of genomic 1,25D-mediated influences on phospholipid composition remains uncertain, potential influences on calcium handling, cell proliferation, and insulin signaling merit further consideration.
G. Bone-muscle cross talk and vitamin D
It is well established that muscle strength and muscle mass are important determinants of bone density, bone geometry, and fracture risk. Vitamin D therefore plays a key role in bone metabolism not only through its direct effects mediated by the VDR in osteoblasts and its effects on calcium absorption by the intestines but also through its effects on muscle fiber size and muscle function noted above. Interestingly, during growth, serum 25D levels have been found to be negatively associated with the accrual of bone mineral content in girls (155), and 25D levels decrease as lean mass increases (156). These observations raise the possibility that muscle tissue may require additional vitamin D during growth and that an important function of vitamin D on bone mass accrual may be mediated by the effects of vitamin D on accumulation of lean mass, which has been documented to precede gains in bone (157).
Another pertinent consideration is the role of FGF23, a protein whose regulation is closely linked to phosphate homeostasis and the activation of vitamin D (step 3, Fig. 2). Although 1,25D up-regulates the expression of FGF23 by osteocytes and osteoblasts, FGF23 inhibits 1,25D synthesis and stimulates its breakdown (158). Therefore, FGF23 excess, as seen in those with oncogenic or X-linked hypophosphatemic osteomalacia, leads to reduced 1,25D and phosphate levels and is also associated with muscle weakness. In Hyp mice, a model of X-linked hypophos-phatemic rickets, the administration of neutralizing FGF23 antibodies increased 1,25D and phosphate levels as well as leading to improvements in grip strength and spontaneous movement (159). Therefore, the FGF23-vitamin D feedback loop presents another layer of complexity when assessing effects on muscle function.
H. Cell models and molecular pathways for insulin signaling and diabetes
I. Insulin signaling in cell models
Treatment of U-937 human promonocytic cells with 1,25D leads to time- and dose-dependent increases in the mRNA expression of insulin receptors, as shown in two separate studies (160, 161). In the second study, 1,25D treatment also resulted in an increase in VDR expression, suggesting that the accompanying increases in insulin-mediated [14C]2-deoxyglucose uptake and [125I]insulin binding in these cells resulted from the activation of genomic pathways. The authors also reported a putative VDRE in the human insulin receptor gene promoter on the basis of luciferase assays on transfected plasmid constructs (162). However, differences in insulin signaling mechanisms between skeletal muscle and immune cells, with the reliance of the latter on GLUT1 rather than GLUT4 transporters for glucose uptake, may limit extrapolation of these results.
In a recent study, differentiated C2C12 muscle cells were rendered insulin resistant and atrophic by treatment with free fatty acids (FFA) (163). However, coadministration of 1,25D with FFA resulted in significant dose- and time-dependent increases in the insulin-mediated uptake of [3H]2-deoxyglucose compared with cells that had received FFA alone. This effect of 1,25D was initially observed at 12 h and reached stability at 36 h, at which point complete reversal of the FFA-mediated insulin resistance was observed (using 10 nM 1,25D). In addition, 1,25D treatment prevented muscle atrophy as demonstrated by significantly increased myotube diameter in cells that had been cotreated with 1,25D and FFA as opposed to those receiving FFA alone.
To account for these effects on insulin resistance, the authors reported that 1,25D treatment reversed a number of FFA-induced abnormalities in the insulin-signaling pathway. At a protein level, 1,25D significantly inhibited FFA-induced serine phosphorylation of IRS-1 and increased the tyrosine phosphorylation of IRS-1 and the phosphorylation of Akt. In addition, the FFA-mediated activation of c-Jun N-terminal kinase, a protein with a significant role in insulin resistance, was significantly reversed by 1,25D treatment. Thus, this report provided the first demonstration of a direct effect of 1,25D in the restoration of key components of the insulin-signaling pathway in an established cellular model of insulin resistance.
2. Arachidonic acid (AA) release
Apart from direct effects on the composition of membrane phospholipids as discussed earlier, there is evidence that 1,25D leads indirectly to the release of AA by a process involving deacylation of membrane-bound phosphatidylcholine (164). This is relevant because levels of AA, a polyunsaturated fatty acid, in skeletal muscle correlate inversely with insulin resistance in humans. AA may also modulate the function of membrane insulin receptors and glucose transporters and may influence the action of insulin by acting as a precursor for the generation of second messengers such as DAG (90). AA also plays a central role in inflammation, with the production of both pro- and anti-inflammatory metabolites that may also have potential effects on insulin signaling.
In chick myoblasts, 1,25D treatment resulted in dose-dependent increases in the release of [3H]AA, but the effect appeared to be dependent on the influx of extracellular calcium and the indirect activation of phospholipase A2 by PKC (164). More research is needed to clarify the possibilities that arise from an effect of 1,25D on AA release and its availability for intracellular processes.
3. Caveolin-I mediated insulin sensitivity
Some data suggests that vitamin D may play a role in caveolin-I mediated insulin sensitivity (165). Caveolin-I, a scaffolding protein within the caveolar membrane, has recently been shown to play an important role in insulin sensitivity. Its selective down-regulation in the skeletal muscle of wild-type mice and its reduced expression in JYD mice, an age-dependent type 2 diabetes model, were associated with significant impairments in insulin sensitivity (166). VDR is present within the caveolae, appear in close proximity to caveolin-1 on confocal microscopy after treatment of ROS 17/2.8 cells with 1,25D (110) and relies on caveolin-I for the mediation of nongenomic effects within skeletal muscle cells (20), all supporting a close association between vitamin D and caveolin-I and raising the question as to whether vitamin D might also have an impact on caveolin-I mediated insulin sensitivity.
Perhaps the strongest evidence to support this possibility is the combination of marked insulin resistance and vitamin D resistance in humans with homozygous nonsense caveo-lin-1 mutations, otherwise known as Berardinelli-Seip congenital lipodystrophy (167). Future research may address this intriguing conceptual link.
IV. Vitamin D and Muscle: Studies in Animal Models
A. VDRKO mice
The VDRKO mouse model has provided valuable insights into the biological function of the vitamin D endocrine system and, specifically, the genomic activity of the transcription factor VDR (13). The role of vitamin D in the development, morphology, and function of skeletal muscle has been made clearer by studies on this mouse model.
The earliest form of the VDRKO mouse, generated by targeted ablation of the DNA encoding the second zinc finger of the DNA-binding domain, was described in 1997 (28). Mice appeared phenotypically normal at birth, despite known expression of the VDR in fetal life in wild-type mice, but became hypocalcemic with secondary hy-perparathyroidism around weaning (21 d) and developed alopecia associated with large dermal cysts by 4 wk of age and rickets and growth retardation by d 35. The initial lack of hypocalcemia is considered to be due to the early presence of nonsaturable 1,25D-independent mechanisms of intestinal calcium absorption. However, when 1,25D-dependent mechanisms take over, mice require a high calcium (2%), phosphorus (1.25%), and lactose (20%) rescue diet for survival. The alopecia is interesting, because people with mutations in VDR (vitamin D-resistant rickets) also develop alopecia (168, 169).
1. Muscle morphology and development
A study in 2003 described histological changes in the muscles of VDRKO mice directly before and after the development of hypocalcemia. At 3 wk of age, samples taken from the quadriceps muscle of VDRKO mice displayed a wider degree of variability in fiber diameter in addition to significant reductions in the size of both type I and II fibers compared with those in wild-type mice (21). By 8 wk of age, generalized atrophy of type I and II muscle fibers had worsened in the VDRKO mice, suggesting progression due to the absence of the VDR or the additional effect of systemic biochemical changes that had not been present at 3 wk. These changes were also reported in VDRKO mice on a high-calcium, -phosphorus, and -lactose rescue diet, suggesting that the absence of VDR was the predominant cause rather than systemic biochemical changes. Neither degenerative nor necrotic changes were observed in VDRKO skeletal muscle, and similar results were obtained in a range of other muscle groups in this model, suggesting a diffuse effect of the VDR on skeletal muscle morphology.
Impaired regulation in the expression of particular myogenic transcription factors, known to control muscle phenotype, was reported as an explanation for these findings. On immunohistochemistry, Northern blot, and RT-PCR analyses, the expression of myf5, myogenin, and E2A was significantly higher in quadriceps samples of 3- and 8-wk-old VDRKO mice compared with age-matched wild-type mice. Persistent expression of immature forms of MHC was also found in the small muscle fibers of VDRKO mice but not in their type II muscle fibers. The expression of two other myogenic transcription factors, namely MyoD and MRF4, were not particularly affected in VDRKO mice.
Although these data support a role for vitamin D in the regulation of muscle development, precise genomic mechanisms by which the VDR influences myogenesis are unclear, and the issue is further complicated by the failure to identify negative VDRE in the promoter region of the genes encoding myf5 and myogenin (170, 171).
2. Muscle strength and functional assessment
A number of studies have examined muscle strength and performance in VDRKO mice. These investigations rarely assess muscle function in isolation, with results being potentially influenced by a range of other factors including cardiovascular endurance, balance, and the ability to learn new skills. Furthermore, behavioral differences between VDRKO and wild-type mice have been reported, which may confound the assessment of some tests (172). Nevertheless, these studies provide an indication of the functional motor deficits associated with loss of VDR by testing swimming ability and motor coordination.
Forced swim test analysis. Three studies have described differences in the swimming behavior of VDRKO mice. In one study comparing six VDRKO mice with 10 wild-type and heterozygote mice that were shaved to account for alopecia-related alterations in buoyancy, severe impairment of swimming was seen in the knockout mice (173). Baseline levels of locomotor, sensory, and vestibular activity were similar. On swim testing, they were observed to swim in a predominantly vertical position, had a significantly greater number of sinking episodes, and displayed stereotypic rotations and catatonic-like upper limb spasms.
B In another study, a significantly greater number of sinking episodes were seen among eight VDRKO mice compared with six wild-type controls (174). However, on being given a stimulus, they showed no impairment in ability to move down a 1-m laneway or in reaching a visible platform. However, VDRKO mice did display greater fatigue after the swim based upon differences in rearing and grooming behavior. The authors hypothesized that this was due to the reduced availability of calcium in VDRKO mice after exercise.
A third study reported that abnormal patterns of swimming behavior among VDRKO mice were more marked in older (5-13 months) rather than younger knockouts (175). Interestingly, no such impairment in swimming ability was seen among a group of 11 1 a-hydroxylase (Cyp27B1)-knockout mice (175). That suggests the intriguing possibility that there might be vitamin D-inde-pendent effects of VDR upon muscle function (175).
B Tests of motor coordination.
The time taken for a mouse to fall off a device that rotates at fixed speeds and in acceleration is considered to indicate its degree of motor coordination (174). In one study, VDRKO mice stayed on a rotating device for a significantly shorter time than wild-type mice on both the accelerating and fixed-speed rotarod tests (174). On gait assessment, VDRKO mice took significantly shorter steps and traveled a shorter distance than wild-type mice when placed in an open field for 5 min.
Significantly shorter retention times on accelerated ro-tarod testing were confirmed in another group of 12-month-old VDRKO mice compared with age-matched wild-type mice (175). These 12-month-old mice demonstrated similar impairments in muscle coordination in tilting box and tilting tube tests, which measure the latency and the angle at which the animal displaces off a device that, respectively, tilts or rotates at different angles. However, no differences were found in 6-month-old VDRKO mice.
In another study, the vertical screen test that measures the time taken for a mouse to fall from a screen that becomes suddenly vertical while the mouse rests on it in a horizontal position (i.e. retention time) was employed (173). VDRKO mice demonstrated markedly shorter retention times compared with both wild-type and heterozygous groups, implying impaired motor coordination or strength.
In summary, VDRKO mice demonstrate notable defects in their overall motor performance with significant impairments in their ability to remain afloat while swimming and shorter stride length and impaired motor coordination and balance on rotarod, rotating tube, and tilting box tests. Although the effect of alopecia was accounted for in one study, other biochemical and behavioral changes in VDRKO mice together with the widespread expression of the VDR in the central nervous system, vestibular system, and spinal cord under normal conditions make direct assessment of its role in muscle function difficult in this setting (175). Nevertheless, it is clear that the overall motor function of these mice is impaired and that further work is needed to clarify the individual components that may account for this.
B. Other animal models
Although the VDRKO mouse model has provided valuable insights into the biological activity of vitamin D, it is more accurate to consider this as a model of type II vitamin D-dependent rickets rather than vitamin D deficiency. Studies have examined muscle function in animals rendered vitamin D deficient by a range of dietary and other methods (176). In a study from 1978, vitamin D-deficient male Sprague-Dawley rats demonstrated significantly prolonged times to peak tension and recovery times in electrically stimulated soleus muscle contraction compared with vitamin D-replete rats on normal chow (177). These changes normalized after vitamin D repletion. In contrast, no impairments were noted in rats rendered phosphate deficient or calcium deficient 10 d before experimentation.
A study from 1979 examined muscle function in chicks raised from hatching on a vitamin D-deficient diet (178). Significant reductions in the tension generated by the triceps surae during stimulation of the posterior tibial nerve compared with that in vitamin D-replete chicks were found. This occurred independently of calcium and phosphate levels. The authors found no difference in the histological appearance of muscle samples but did isolate reduced muscle mitochondrial calcium levels. This was independent of serum calcium levels and was proposed as the mechanism by which vitamin D deficiency affected muscle contraction force.
Four weeks after the commencement of a vitamin D-deficient diet, rats displayed marked skeletal muscle hypersensitivity on calf compression compared with those fed a normal diet (179). This finding was not related to hypocalcemia but was rather accelerated by increased dietary calcium and was accompanied by early impairments in balance on a beam-walk test to assess the frequency of foot slips. Histologically, vitamin D-deficient rats displayed increased numbers of presumptive nocioceptor axons in skeletal muscle, providing an explanation for their hyperalgesic phenotype.
In contrast to these studies, a recent report questioned the primary role of vitamin D, rather than biochemical changes associated with vitamin D deficiency, in resulting in myopathy (176). In 58 male Wistar rats that were rendered vitamin D, phosphorus, and calcium deficient by dietary methods and housing under incandescent lighting, muscle strength of the soleus as assessed by a force transducer detecting isometric contraction was significantly reduced compared with that in replete controls (176). The authors concluded that phosphorus deficiency was the primary culprit on the basis of several findings. These included a direct independent correlation between phosphorus levels and the reduction in soleus muscle force in these animals, complete restoration in slow-twitch muscle force after dietary repletion with phosphorus despite persistent vitamin D deficiency and similar measures of muscle function among phosphorus-replete rats that were either vitamin D deficient or replete. Similarly, there was no difference in muscle contraction among rats that were calcium deficient and replete, in which vitamin D and phosphorus levels were within the reference range. The central importance of phosphorus in the production of ATP, essential for muscle contraction, was considered as the explanation for these findings.
Therefore, the difficulty in differentiating the effects of severe vitamin D deficiency from those of hypocalcemia and hypophosphatemia in the development of muscle pathology is common to animal and human clinical studies. However, cell lines and tissue culture enable study of direct effects, and these studies date back to the 1970s.
C. Animal studies on insulin sensitivity and diabetes
Although vitamin D deficiency has been associated with more aggressive disease among nonobese diabetic mice (180), a model for type 1 diabetes, few animal trials have addressed the role of vitamin D in insulin resistance. In one study of ob/ob mice, an obese type 2 diabetes model, significant improvements in hyperglycemia and hyperinsulinemia in response to treatment with 1a-hydroxyvitamin D3 (1a-OHD3) were observed (181). In another study of aged rats with type 2 diabetes and insulin resistance that received 25D, 1a-OHD3, or no treatment, serum 1,25D levels correlated positively with the glucose infusion rate on euglycemic clamp studies at the end of the 12-wk treatment period (182), suggesting a potential role in insulin sensitivity. However, the main focus of this study was the demonstration of bone loss among insulin-resistant rats attributed to a reduction in their renal 1a-hydroxylase activity.
D. Summary: vitamin D and muscle function in animal studies
Congenital absence of the VDR in VDRKO mice is associated with atrophy of both type I and type II fibers, poor musculoskeletal performance in behavioral tests, and marked changes in gait. Vitamin D deficiency induced in rats has also been found to alter muscle function when compared with vitamin D-replete animals. Together, these studies using animal models provide additional evidence that vitamin D plays an important role in muscle physiology. However, they do not separate the effects of muscle vs. whole-body deletion of VDR. Biochemical abnormalities in these mouse models may once again confound these findings (Fig. 4). A similar question has been recently answered in relation to cardiac muscle; mice with specific deletion of VDR in cardiac muscle displayed significant cardiac hypertrophy, similar to that seen in whole-body VDRKO mice (183). Specific deletion of VDR in skeletal muscle using Cre-Lox technology will be required to clarify this question.
V. VDR Polymorphisms and Muscle Function
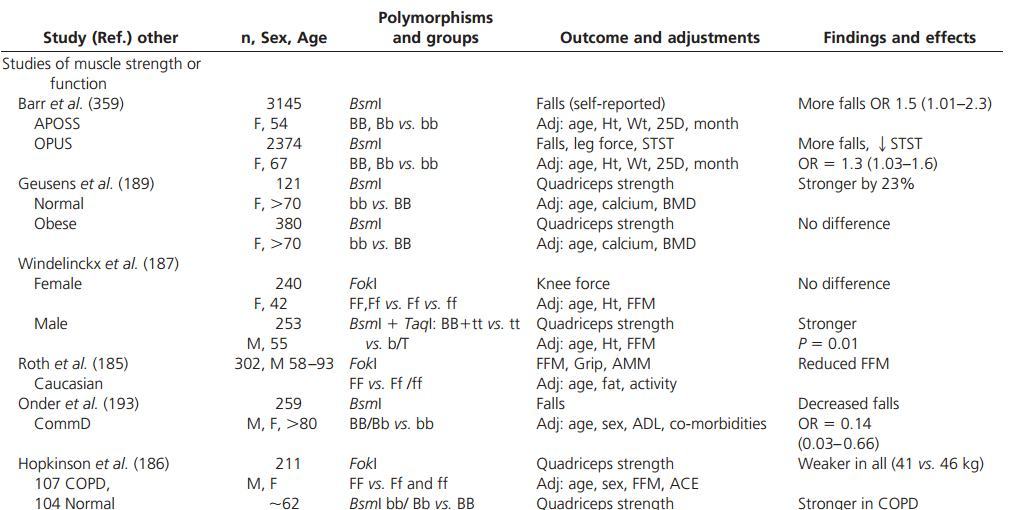
Several single-nucleotide polymorphisms (SNP) in the gene encoding the VDR have been associated with a range of phenotypic characteristics including muscle strength. These have been summarized in Table 3.
A. FokI polymorphism
The FokI polymorphism of the VDR gene is a T/C transition in exon 2 of the VDR gene that results in a shorter 424-amino-acid protein with enhanced transactivation capacity using a reporter gene assay (184). Although this greater VDR activity might suggest improved muscle strength in light of some of the clinical data, the FokI polymorphism is associated with reduced muscle strength in two studies in men (185, 186). It is interesting to speculate that increased VDR function could increase CYP24A1 expression and thereby degradation of 1,25D.
Among 302 Caucasian men (aged 58 -93 yr), those who were homozygous for the FokI polymorphism displayed significantly lower fat-free mass and appendicular muscle mass on dual-energy x-ray absorptiometry (DEXA) scanning (185). Furthermore, men with this polymorphism demonstrated a 2.17-fold higher risk of sarcopenia (defined by ap-pendicular fat-free mass <7.26 kg/m2), independent of age. Lower fat-free mass was associated with significantly lower quadriceps muscle strength. Differences in quadriceps strength were not significant after adjusting for fat-free mass, suggesting that the difference was mediated by altered muscle mass.
In another study that included 107 patients with stable chronic obstructive pulmonary disease and 104 healthy, age-matched controls, homozygosity for the FokI polymorphism was associated with reduced quadriceps strength compared with heterozygosity or control subjects (186). The difference became more significant in a model that adjusted for age, sex, forced expiratory volume in 1 sec (FEV1), fat-free mass, and angiotensin-converting enzyme genotypes. There was no evidence that the presence or absence of lung disease affected the relation between FokI genotype and quadriceps strength.
Among 240 women (41.5 ± 13.2 yr), those with the FokI polymorphism (FF) had weaker isometric knee extensor strength (P < 0.05 for both 90° and 120° incline) vs. those who were heterozygous (Ff) or lacking the polymorphism (ff) (187). However, on adjusting for age, height, and total fat-free mass, the differences were no longer significant, again suggesting that the polymorphism may be affecting muscle mass.
B. BsmI polymorphism
The data are less consistent for the BsmI polymorphism. This SNP is located in the 3' region of the VDR gene, known to play an important role in the regulation of gene expression (188). In a group of 121 nonobese, healthy women over 70 yr of age, the bb genotype was associated with 23% higher quadriceps strength and 7% higher wrist strength compared with those with the BB genotype (189). However, among the 380 obese women over the age of 70 in this same study, no effect was found.
Conversely, a study of 175 young healthy women (age range 20 -39 yr) in Belgium found that those with the bb genotype had lower hamstring strength on dynamometer and lower fat-free mass on DEXA compared with the BB genotype (190). The difference in hamstring strength became of borderline significance after adjustment for age, fat mass, and lean mass. Interestingly, the significance of another polymorphism known to be in linkage disequilibrium with the BsmI B allele, namely the polyadenosine repeat (ss genotype), was associated with higher hamstring strength and greater body weight and fat mass compared with those with LL genotype in this study.
Among 109 healthy female university students in China (age around 19-21 yr), those with the bb genotype of the BsmI polymorphism displayed significantly lower peak torque in concentric knee flexors at a specific setting on the isokinetic dynamometer than the combined BB and Bb group (P = 0.03) (191). However, when this parameter was tested at other settings, namely 120°/sec and 30°/sec, the differences were not statistically significant.
TABLE 3. (just the top)
When examining another polymorphism at the ApaI site, peak torque in eccentric knee
Studies assessing the relation between VDR polymorphisms, falls, muscle function, and markers of insulin 2374 extensors at 120°/sec was significantly lower in the AA homozygous group compared with the aa and Aa groups.
In a study of 253 men (54.9 ± 10.2 yr), Bt homozygotes ( i.e. those with BB genotype at the BsmI site and tt genotype at an associated polymorphism site, namely TaqI) had higher isometric quadriceps strength at 150° on isokinetic dynamometer than b or T allele carriers without and with adjustment for confounding factors (P = 0.01 after adjustment) (187). However, no such association was found when this same parameter was tested at 90° or 120° or when assessing knee flexor strength.
Among 120 Turkish men (>65 yr), knee extensor strength on dynamometer was significantly higher in those with BB homozygosity at the BsmI site than in the Bb/bb group, but no significant association between muscle mass and strength was found (192).
Two population studies examining the rate of falls suggest that the bb genotype of the BsmI polymorphism may be protective against falls. In a study from Italy that included 259 community-dwelling older patients (>80 yr of age), the rate of falls differed according to BsmI genotype with more seen among those with BB or bb genotype on multivariate analysis (193).
Data collected from two separate population cohorts of older women, namely the Aberdeen Prospective Osteoporosis Screening Study (APOSS) and Osteoporosis and Ultrasound study (OPUS), also identified a greater incidence of self-reported recurrent falls among those with the BB genotype of the BsmI polymorphism compared with those with the bb genotype (194). Significant differences in function with greater ease in rising from a chair were seen in bb ho-mozygotes compared with carriers of the B allele. These studies failed to demonstrate an association between Fok1 polymorphisms and balance or muscle power measurements.
Apart from VDR polymorphisms, a recent population-based study that involved 153 men and 596 women (65101 yr) reported an association between SNP in the CYP27B1 gene (i.e. -1260 and +2838) and the risk of fracture over a 2.2-yr follow-up period (195). There was no difference in the risk of falls among subjects, and muscle strength was not examined. There are limitations in the interpretation of these data. Larger studies to assess the association between muscle strength and genetic polymorphisms are needed, and more functional studies of effects on VDR function are required.
C. VDR polymorphisms and insulin resistance/type 2 diabetes
Particular polymorphisms in the gene encoding the VDR may be associated with the development of insulin resistance and type 2 diabetes among certain populations. However, once again, the data are inconclusive due to the generally small sample size in these studies and the variability in the populations and examined endpoints. These studies have been summarized in Table 3.
VI. Vitamin D and Muscle: Human Studies
A. Myopathy
Rickets and osteomalacia have been associated with muscle weakness and hypotonia for centuries (196, 197). Weakness affecting the proximal lower limb musculature was reported in a group of adults with osteomalacia who responded to high-dose vitamin D therapy in the 1960s (198). The paper did not report whether all patients responded or whether responses were complete.
In addition to general weakness, more specific proximal muscle deficits are commonly described, including difficulty rising from a seated or squat position, ascending a flight of stairs, or lifting objects (199-201). Changes in gait, often described as waddling or penguin-like in appearance, are widely reported and are possibly a combined result of bone pain, muscle pain, and proximal weakness (199, 202). Pictures of a child with rickets demonstrates the potentially profound effects of vitamin D deficiency (Fig. 7). However, the classic pattern of proximal weakness seen in vitamin D deficiency is not specific. Many endocrine and metabolic disorders including renal failure, hyperparathyroidism, hypophosphatemia, Cushing's syndrome, and hyperthyroidism as well as glucocorticoid therapy may display similar clinical features (203, 204). Electromyographic changes seen in vitamin D-deficient subjects with muscle weakness confirm myopathy, but without specific features (201). In the reports of myop-athy with vitamin D deficiency, many subjects had multiple biochemical abnormalities involving calcium, phosphate, and PTH that co-corrected with vitamin D repletion, making it difficult to assess the individual role of each component in the development of osteomalacic myopathy (205). These observations formed the basis for the belief that myopathy in these subjects was not directly related to vitamin D deficiency but rather a general result of osteomalacia and its associated biochemical abnormalities (206).
Observational and uncontrolled treatment studies
The reversibility of myopathy with vitamin D supplementation has been described in some case series (200, 207, 208). A recent series described the presence of progressive muscle weakness among young vitamin D-defi-cient veiled women from Saudi Arabia [90% had 25D < 8 ng/ml (20 nmol/liter)] (205). Some women required a wheelchair.
Figure 7. (Just C&D;)
Figure 7. Images of a child with rickets. These images of a 2-yr-old child with rickets display widening of the metaphyseal regions in the left wrist (A) and both ankles (B). The x-ray of the left wrist shows widening of the radial and ulnar metaphases with typical moth-eaten appearance of the physis and rarefaction of the metacarpal and phalangeal bones (C) with subsequent improvement after 3 months of treatment with cholecalciferol (D). These bone deformities are due to impaired mineralization of bone at a critical stage in its development. Muscle weakness, wasting, and hypotonia are also widely reported features of rickets, but their mechanism is unclear. Images are courtesy of Associate Professor Craig Munns, Childrens' Hospital Westmead, Sydney, Australia.
Substantial improvements followed 3 months of vitamin D and calcium supplementation (800 IU and 1200 mg daily, respectively). Wheelchair-bound patients walked independently by the end of the study.
In another case series, five patients with myopathy resulting in wheelchair use were treated with vitamin D2 (50,000 IU weekly) (209). At baseline, they were deficient [25D = 5-13 ng/ml (12-32 nmol/liter)] with secondary hyperparathyroidism (intact PTH range = 13-89 pmol/ liter). Treatment resulted in marked improvements in strength, pain, and mobility within 4 -6 wk, despite persisting hyperparathyroidism. The authors suggested that this indicated a role for vitamin D independent of PTH.
A study assessing vitamin D-deficient women [25D = 7 ng/ml [17 nmol/liter)] vs. controls with higher levels [25D = 19 ng/ml (47 nmol/liter)] reported an independent association between 25D levels and maximal voluntary knee extension force (208). No correlations were found for PTH or total or bone-specific alkaline phosphatase.
A common theme in these case series that describe patients with muscle pain and weakness is the high rate of initial misdiagnosis. The diagnoses of diabetic neuropathy, general debility, motor neuron disease, orthopedic disorders, psychiatric conditions, or inherited myopathy were described before the recognition of vitamin D deficiency (205, 209, 210). The nonspecific clinical features of vitamin D-deficiency myopathy, the wide range of severity from mild weakness to debilitating pain and immobility, and low index of suspicion may contribute to the frequent delay in the diagnosis.
We are not aware of any reports of muscle biopsy or muscle function studies in people with mutations in VDR.
B. Myalgia and vitamin D deficiency
People with vitamin D deficiency and proximal myopathy (i.e. weakness of the proximal musculature arising from muscle pathology) often have associated proximal myalgia (i.e. muscle pain) (198). Many authors have also proposed that low vitamin D is associated with more diffuse muscle pain (211-213). However, this is controversial, and other studies do not support this (214 -216). The issue is made more difficult because osteomalacia is associated with bone pain and microfractures, making causal discrimination of the source of pain challenging. Because it is conceptually obvious that people with muscle pain may be less likely to exercise, go out, and carry out normal outside activities of daily living, establishing cause and effect is important. Ideally, demonstrating a therapeutic response would clarify the issue.
1. Observational studies
A cross-sectional study of 3075 men from eight European centers found that those who reported chronic widespread pain (8.6%) were more likely to have low 25D levels [<15 ng/ml (37.5 nmol/liter]) (217). However, the relationship was attenuated by adjusting for age, season, activity, and other factors.
Ninety-three percent of 153 patients who were being assessed for persistent, nonspecific musculoskeletal pain in Minneapolis were vitamin D deficient [mean 25D = 12 ng/ml [30 nmol/liter)] (211). All African-American, East African, Hispanic, and American Indian patients had 25D below 20 ng/ml (<50 nmol/liter) as did 82% of Caucasian patients. None had fibromyalgia or medical conditions known to decrease production, absorption, or hydroxy-lation of vitamin D. This study did not include a control group of similar ethnicity.
The propensity for particular ethnic groups, including those migrating from Asia and the Indian subcontinent to Western countries, to develop myalgia and bone pain as the primary manifestation of vitamin D deficiency has been reported since the 1970s (218,219). A study describing 33 mostly Somalian female asylum seekers with mus-culoskeletal pain and low 25D [<8.5 ng/ml [21 nmol/ liter)] found that vitamin D and calcium supplementation led to the symptom resolution in 22 (66.7%) by 3 months (220). The authors noted a 2.5-yr mean lag time between symptom onset and diagnosis. There was no control group. A study from the United Arab Emirates found that 86% of patients who were initially diagnosed with fibro-myalgia or nonspecific muscle pain were vitamin D deficient. The majority reported improvement in response to supplementation (221). Another study compared cultural differences in the reporting of muscle pain among South Asians (i.e. Indian, Pakistani, and Bangladeshi) and white Europeans living in England (212). Reporting of widespread pain was significantly more common in the 1945 South Asians. However, in the 137 South Asians in whom 25D was measured, there was no association between deficiency and pain.
A report describing myalgia in six women who had migrated to the Netherlands considered vitamin D deficiency [<8 ng/ml (<20 nmol/liter) in 5 patients] to be the cause. There was a lengthy lag period (7-103 months) from the onset of symptoms to diagnosis (222). In three cases, misdiagnosis led to treatment with prednisone, estrogen, or cholecystectomy. Vitamin D and calcium supplementation was effective in reversing the myalgia in each case.
2. Case-control studies
Muscle pain was recently proposed as a marker for vitamin D deficiency among Aboriginal Australians (223). A case-control study of eight urban Aboriginal patients with muscle pain and eight matched Aboriginal controls without pain reported significantly lower vitamin D levels among those with pain [17 vs. 23 ng/ml (41 vs. 58 nmol/ liter), P = 0.017].
Wide cultural and gender differences in the reporting of pain, subjective features in the diagnosis of fibromyalgia, and the presence of other features known to affect vitamin D status among patients with persistent pain syndromes confound the assessment of the role of vitamin D deficiency in muscle pain. Also, the observational nature of these studies does not equip them to address the question of causality.
3. Randomized controlled trials (RCT) for myalgia
We identified only one randomized placebo-controlled trial of supplementation where treatment of muscle pain appeared to be the primary endpoint. The study examined people with diffuse muscle pain (214). Fifty subjects with 25D below 20 ng/ml (50 nmol/liter) at baseline were randomized to placebo or vitamin D2 50,000 IU weekly for 3 months. There was no benefit of treatment. The authors note that 50% of the placebo group achieved normal vitamin D levels during the study; however, the improvement in pain scores was not substantial for either group. Using the PowerStat program (224), we calculate that the study had 80% power with a of 0.05 to detect a difference of ± 19 in pain score. The baseline visual analog pain score in the treatment group was high at 67 ± 23, so if there is any benefit, it is probably smaller than this.
4. Summary: myalgia
Vitamin D deficiency with osteomalacia is associated with muscle pain that in most cases resolves with treatment. The pain is more commonly located in large proximal muscle groups rather than displaying diffuse distribution (198, 222, 225, 226) and is often associated with bone pain and other features of osteomalacia and myopathy (205, 208).
Thus, for pain (proximal or diffuse) in patients without osteomalacia, other etiologies should also be considered. In patients with diffuse pain, without obvious osteomalacia, the data remain inconclusive. There are strong associations, but the only RCT found no convincing benefit of supplementation. The trial was adequately powered to detect a clinically meaningful change in pain score.
There are no RCT examining specific treatment of proximal muscle pain. Larger randomized placebo-controlled trials should be carried out, preferably with stratification by baseline vitamin D status, in people with fibromyalgia and in people with proximal myalgia. There is no evidence to support supplementation for myalgia in people with normal levels.
C. Fibromyalgia
Fibromyalgia is not purely a muscle disorder, but there are a number of studies examining the potential association between it and vitamin D. Myalgia was examined in a cross-sectional study of 6824 white, middle-aged subjects living in the United Kingdom. A significant association between fibromyalgia, defined by the American College of Rheumatology criteria, and 25D was found among women (227). However, no such association was found in men, although they reported similar rates of pain (11.4% for men, 12.5% for women). No association with myalgia was found.
In two separate case-control studies of patients with fibromyalgia from the United States and Brazil, no statistically significant differences were found in 25D levels (214, 228).
In contrast, another case-control study that examined 40 premenopausal women with fibromyalgia and 37 controls found a significantly higher proportion of vitamin D deficiency [25D < 8 ng/ml (20 nmol/liter)] among women with fibromyalgia (229). However, this was not adjusted for physical activity, smoking, or body mass index (BMI). Fibromy-algia symptoms did not differ depending on the vitamin D status. A subset of deficient patients who received supplementation (eight of 18) reported subjective improvement that persisted at 3 months; however, 10 of 18 did not.
One placebo-controlled study examining the effect of vitamin D on fibromyalgia was identified (230). In that study of 138 patients with fibromyalgia, the subset of 100 patients with mild to moderate vitamin D deficiency and insufficiency [25D = 10-25 ng/ml (25-62.5 nmol/liter)] who were randomized to receive vitamin D3 (50,000 IU weekly) showed significant improvement over 8 wk vs. placebo-treated controls. However, this did not persist at 1 yr, and in the same study, the subset of 38 people with severe deficiency [25D <10 ng/ml (25 nmol/liter)] who received vitamin D in an unblinded fashion did not report any improvement at either 8 wk or 1 yr (230).
Overall, there is conflicting evidence regarding the possibility of an association between low vitamin D and fibromyalgia, and clearly, having fibromyalgia may have an impact on time spent outdoors/exercising. The data from the one randomized placebo-controlled trial do not provide a clear answer, because the most deficient subjects did not benefit, but less deficient subjects did. More research is needed in this area.
D. Drug-related myopathy and vitamin D
1. Aromatase inhibitors
The effect of vitamin D supplementation on myalgia due to a drug class, namely aromatase inhibitors, has been recently reported. For this class, there is both observational and randomized controlled data, and myalgia is a very common side effect.
An observational study found significantly less musculoskeletal pain among 60 women on letrozole who had achieved median 25D levels over 66 ng/ml (165 nmol/liter) compared with women with levels below 66 ng/ml (165 nmol/liter) (19 vs. 52%) after weekly supplementation with 50,000 IU vitamin D3 for 12 wk (231).
In a double-blind RCT, 60 patients with early-stage breast cancer with new or worsening musculoskeletal pain on aromatase inhibitor therapy were randomized to receive either high-dose vitamin D supplementation at a regimen that depended on their baseline 25D or placebo (232). Those with 25D of 10-19 ng/ml (25-47 nmol/liter) received 50,000 IU vitamin D2 for 16 wk, whereas those with 25D of 20-29 ng/ml (50-72 nmol/liter) received 50,000 IU for 8 wk (232). There were significant improvements in pain at 2 months in the vitamin D group vs. placebo on the basis of several indices. Therapy was decreased from weekly to monthly after 2 months in most subjects. The beneficial effects did not remain at the 4- and 6-month visits. There was evidence of a dose-response effect; women who were more deficient had greater benefits, and in that subgroup, the beneficial effect was seen across the whole study period.
2. 3-Hydroxy-3-methyl-glutaryl-coenzyme A reductase inhibitors (statins)
A number of reports suggest that vitamin D deficiency may potentiate myopathy in patients on lipid-lowering statin therapy (3-hydroxy-3-methyl-glutaryl-coenzyme A reductase inhibitors) (233-242). These reports are both anecdotal and based on case series and cross-sectional studies. Not all studies have confirmed this relationship (237).
Cross-sectional and cohort studies.
Among a group of 621 statin-treated patients, the 128 who reported myalgia had lower serum 25D (238). Levels were low [defined as <32 ng/ml ( < 80 nmol/liter) in that study] in 64% of myalgic patients vs. 43% of the others. A subset of 38 of the 82 25D-deficient myalgic patients were treated with 50,000 IU/wk of vitamin D2 for 12 wk (unblinded) while continuing statins, and 35 had their myalgia resolve. The three nonresponders achieved similar vitamin D levels.
A prospective study of 150 statin-intolerant patients with 25D below 32 ng/ml (80 nmol/liter) reported that 83% were free of myalgia and no longer statin intolerant after approximately 8 months of vitamin D2 treatment (50,000 IU twice weekly for 3 wk and then 50,000 IU weekly) (240). However, treatment had little effect on elevated creatine phosphokinase (CPK).
However, not all studies find an association. A retrospective study of 6808 statin users found no correlation between 25D levels, myalgia, or CPK levels (242). A smaller study of 129 patients on statins also found no difference in the 25D levels of those with and without myalgia (237).
Some hypotheses have been proposed regarding mechanisms by which vitamin D deficiency may potentiate statin-induced myalgia. The suggestion that statins may reduce vitamin D synthesis by inhibition of synthesis of its cholesterol precursor has not been supported by clinical studies (243, 244). The enzyme CYP34 demonstrates in vitro 25-hydroxylase activity (245), and it has been speculated that vitamin D deficiency may lead to preferential shunting or use of this enzyme for vitamin D hydroxylation, reducing its availability to metabolize (deactivate) statins (239). This hypothesis could be tested by measuring statin levels in vitamin D-deficient and vitamin D-suf-ficient individuals matched for the presence or absence of myopathy and/or CPK levels.
Although an association between a polymorphism of the solute carrier organic anion transporter family member 1B1 gene (SLCO1B1) and statin-induced myalgia was reported, no interaction between vitamin D deficiency and genotype was found in a group of 46 patients on statin therapy (241).
These studies of statin myalgia/myopathy are interesting; however, their unblinded nature and lack of control groups are particular limitations, especially given the well-known placebo effect in pain studies. At this time, the evidence of an association between vitamin D deficiency and statin-related myalgia is derived from observational studies and small series. Obviously, people who experience pain with muscle movement may be less likely to be participating in outdoor activities and may therefore make less vitamin D than their pain-free counterparts. Randomized placebo-controlled studies are needed. In the meantime, we suggest treating deficient patients because of the known bone and calcium benefits. There is at present no evidence to support treating people on statins who have normal vitamin D status.
3. Summary: drug-related myopathy and vitamin D
Myalgia has been observed in patients receiving statins or aromatase inhibitors. A number of these studies reveal that patients on statins who suffer from myalgia are also vitamin D deficient, and vitamin D supplementation might improve symptoms of myalgia in these patients. These findings suggest that vitamin D deficiency may potentiate statin-induced myalgia, but the molecular mechanisms underlying the interaction between vitamin D, statin treatment, and muscle function have not been elucidated. Based on the limited data, we recommend treating all deficient subjects who are receiving statins or aromatase inhibitors. The only RCT in patients taking aromatase inhibitor used high-dose therapy. If this is used, patients should be monitored for hypercalcemia and hypercalciuria.
E. Falls and vitamin D
Approximately 30% of community-dwelling people over the age of 65 fall each year, and approximately 20% of these require medical attention (246). Falls pose a substantial risk to an aging population, are a major risk factor for fracture and other injury, and impact negatively on quality of life (247). Therefore, identifying reversible factors is important. In older individuals, falls are closely related to sarcopenia (loss of muscle mass), loss of muscle tone, and a range of conditions that contribute to the complex syndrome of frailty (248). This issue has also recently been discussed in an Endocrine Society statement on ex-traskeletal roles of vitamin D (12).
1. Falls: observational studies
There is a seasonal variation in the incidence of falls, with more occurring in winter among older women. This raises the suggestion of a putative role for vitamin D in the occurrence of falls (249), although other factors such as decreased daylight and increased likelihood of slipping on wet or icy surfaces could also explain the differences. The possibility that vitamin D deficiency, which is highly prevalent in frail and older individuals, may contribute to falls has been examined by a number of studies. These are summarized in Table 4.
In 1619 women in low-level and high-level residential care (mean age 84 yr), a significant inverse association between serum 25D levels and the incidence of falls over approximately 5 months was reported. The authors estimated a 20% reduction in falls risk with doubling vitamin D status on analysis of log[25D] levels (250). A similar-sized study of 1231 community-dwelling individuals identified baseline 25D as a predictor for falls over a 1-yr period, particularly among those 65-75 yr of age (251). Those with 25D levels over 10 ng/ml (25 nmol/liter) displayed the highest risk of recurrent falls.
However, in a smaller study, although older individuals who fell had significantly lower 25D levels vs. the overall group of 83 subjects, this was not significant on multivariate analysis (252).
The possibility that 25D does not directly contribute to falls but is rather an associative marker of frailty remains a potential limitation in the interpretation of these observational data. As in the situation with myalgia, it is obvious that people who fall may spend less time outdoors and thereby have lower exposure to solar UV radiation. The other issue is that malnutrition is very common in elderly persons, affecting up to 40% of those living in institutions,
TABLE 4.
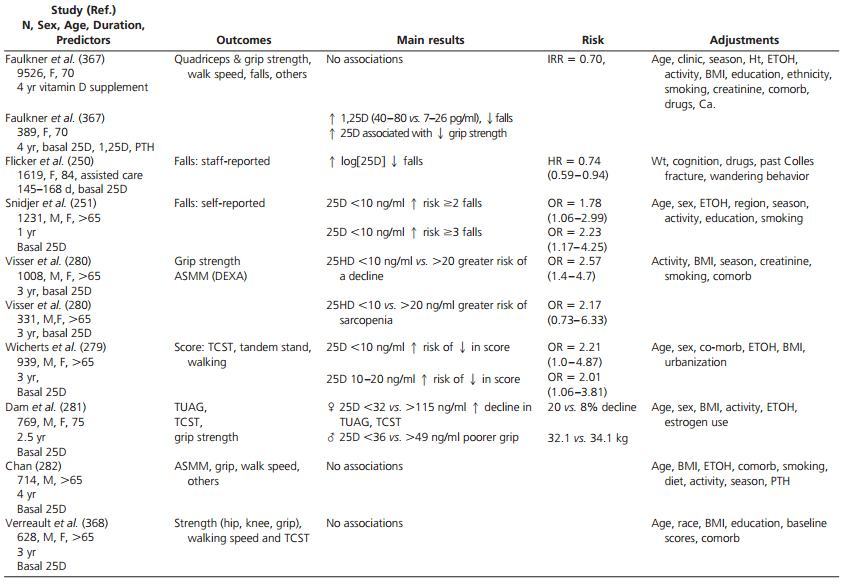
Observational studies assessing the longitudinal effect of vitamin D levels on falls/muscle function (listed in order of number of study participants, highest to lowest)
and may contribute simultaneously to vitamin D deficiency and frailty.
2. Falls: interventional studies
Interventional studies seeking to examine the effects of vitamin D supplementation or UV exposure on the risk of falls among older individuals have been performed and are summarized in Table 5.
a Sunlight therapy.
In a study of 602 residents of aged-care facilities, those who complied with a daily regimen of increased sunlight exposure had fewer falls than those randomized to the control group (253). Compliance was surprisingly poor despite the use of sunlight officers who visited the facilities (median adherence was 26%). Thus, on intention-to-treat analysis, the study was underpowered, and no effects were seen. The authors concluded that vitamin D supplementation was a more practical approach to reduce falls in residential care. It should also be noted that sunlight exposure during the early morning hours rather than at midday when UV levels are highest might not have been sufficiently effective in the synthesis of vitamin D.
b Intervention studies: non-placebo controlled.
The combination of daily 400 IU vitamin D3 plus 1000 mg calcium reduced severe falls requiring admission in 5063 community-dwelling city residents above the age of 66 yr in Denmark (254). The number needed to treat was nine.
c Intervention studies: placebo controlled.
A reduced incidence of falls, regardless of baseline 25D, was seen in a randomized trial of vitamin D supplementation (255). Among 625 older residents of assisted-living facilities, those who received calcium (600 mg daily) and vitamin D2 (initially 10,000 IU once weekly and then 1,000 IU daily) for 2 yr had a lower rate of falls compared with those receiving calcium alone. However, the rate of ever falling was not different, implying that the benefits were among
TABLE 5. (just the top portion)
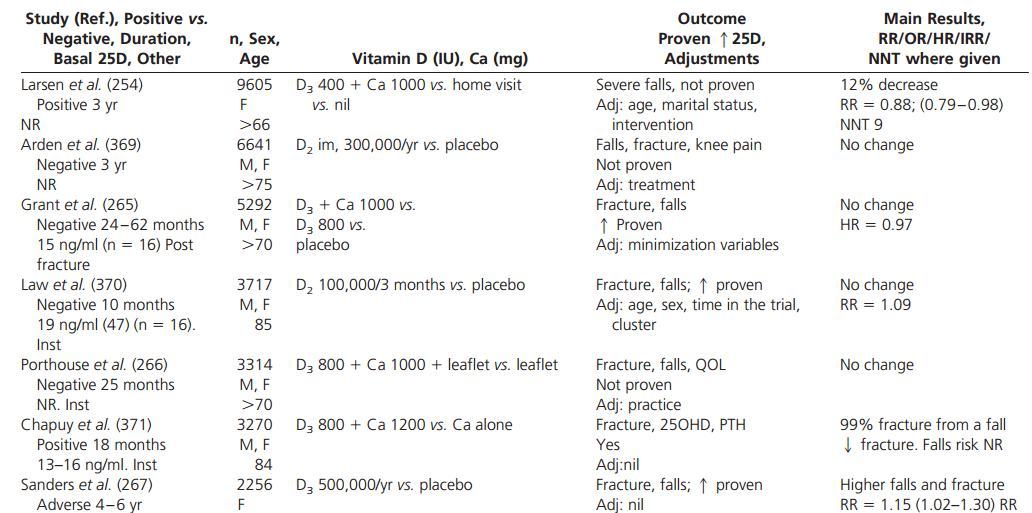
Doses for vitamin D and calcium are daily and oral unless stated otherwise. Adj:, Adjusted for; AGU, acute geriatric unit; BI, Barthel index; CCMI, Charlson comorbity index; CrCl, creatinine clearance (ml/min); E/P, estrogen plus progesterone; ETOH, alcohol use; F, female; HR, hazard ratio; Ht, height; Inst, institutionalized; IRR, incidence rate ratio; ITT, intention-to-treat analysis; M, male; NA, not applicable; NH, nursing home; NR, not reported; OR, odds raito; PASE, physical activity scale for the elderly; QOL, quality of life; RR, relative risk; Wt, weight.
Interventional studies assessing the effects of vitamin D supplementation on the incidence of falls (listed in order of number of study participants, highest to lowest) repeat fallers. On subgroup analysis of compliant participants (defined as taking >50% of the doses, n = 540) the risk of ever falling was also significantly lower. The number needed to treat to prevent one fall per year was eight, similar to the above open-label study.
A 3-yr double-blinded randomized study examined combined supplementation with vitamin D3 (700 IU) and calcium citrate malate (500 mg) vs. placebo in 445 community-dwelling older individuals (256). By intention-to-treat analysis, combined therapy was not effective. A subgroup analysis of the women found a significant reduction in the incidence of falls that was most pronounced in those who were less active. Inter-gender differences in muscle mass were postulated to lead to greater susceptibility for falls among this particular group. The trial was not originally powered to detect effect modification by sex and activity levels, and falls were also a secondary outcome. The primary outcome examined bone density, which was significantly improved (257). In another study, 242 community-dwelling older individuals were randomized to receive calcium (1000 mg daily) with or without vitamin D3 (800 IU daily) for 1 yr and then followed 8 months without treatment but continuing blinding to treatment (258). On intention-to-treat analysis, subjects on dual supplementation reported 27% fewer falls at 1 yr and 39% fewer falls at 20 months.
Among 122 older women in long-stay geriatric care who received calcium (1200 mg/d) with or without vitamin D3 (800 IU/d), those on dual supplementation had significantly fewer falls during the 12-wk treatment period compared with the preceding 6-wk observation period. There was no difference in the proportion who fell at all, but people who fell the most appeared to obtain the greatest benefit (259).
In a particularly vulnerable group, namely older individuals with poststroke hemiplegia, a significant reduction in the number of falls per person and the total number of repeat fallers was seen among 48 women receiving vitamin D2 (1000 IU daily) vs. controls who received placebo for 2 yr (85). The study included biopsies of the unaffected side and reported that the baseline proportion and diameter of type II muscle fibers were significant independent predictors for falls over the 2 yr. In demonstrating significant improvements in these parameters in a subset of individuals who received vitamin D supplementation for 2 yr, they offered a mechanistic explanation for the reduction in falls.
By contrast, recurrent fallers were the least likely to benefit from supplementation in another study (260). Community-dwelling ambulant older women were randomized to receive calcium citrate (1000 mg daily) and either vitamin D2 (1000 IU) or placebo for 1 yr. After adjusting for height, which was not equal at baseline, the vitamin D group had a 19% lower risk of a single fall compared with controls. The effect was most pronounced in winter/spring rather than in summer/ autumn. It was calculated that 25D levels over 22 ng/ml (54 nmol/liter) were adequate for the benefit. The results were not statistically significant without height correction.
Another study that specifically targeted vitamin D-insufficient older individuals [25D <20 ng/ml (50 nmol/ liter)] reported that 800 IU vitamin D3 and 1200 mg calcium daily decreased falls per subject compared with the 148 controls who received calcium alone (261). One non-compliant individual was excluded from analysis. The authors propose that the mechanism relates to a significant improvement in body sway at 8 wk (261, 262). Sagittal body sway improved with treatment, but frontal sway improved in both groups (261).
d. Negative randomized studies and falls. A number of randomized studies have also failed to demonstrate an effect of vitamin D supplementation upon falls.
Among 354 older persons in The Netherlands who were randomized to receive vitamin D3 (400 IU) or placebo for 28 wk, impaired mobility was the major predictor of falls, and vitamin D treatment had no significant effect (263). This study had marked patient heterogeneity with inclusion of both institutionalized and community-dwelling persons.
A positive effect of vitamin D supplementation on falls risk was reported by Broe etal. (264) among the subgroup of 124 nursing home residents receiving high-dose vitamin D (800 IU/d) but not in the 600-, 400-, or 200-IU groups. The risk of falling was lower if those receiving 800 IU vitamin D2 were compared with the combined group of smaller doses (200, 400, or 600 IU) and placebo. There was no suggestion of a normal dose-response curve with the lower doses actually having nonsignificantly higher fall rates compared with placebo (0 IU, 44%; 200 IU, 58%; 400 IU, 60%; 600 IU, 60%; 800 IU, 20%).
A large study of 5292 subjects over 70 yr of age with a recent minimal-trauma fracture did not find a beneficial effect of 800 IU of vitamin D3 on falls over 26-62 months (265). Falls were a secondary endpoint with information collected only for the week before each 4-monthly questionnaire. Compliance (patients taking 80% or more of tablets) was poor (<45% at 2 yr); however, there was also no benefit if only compliant patients were examined. Baseline levels were measured in approximately 1% of patients (n = 60) and were 15.2 ng/ml (38 nmol/liter).
Another study in 3314 older women living in nursing homes did not find fewer falls among those randomized to receive a combination of vitamin D3 (800 IU), calcium (1000 mg), and a falls prevention leaflet over 25 months compared with those receiving the leaflet alone (266). Falls were also a secondary outcome in this study.
In reaction to generally poor adherence to daily regimens of vitamin D supplementation as described by a number of these reports, other studies have examined the efficacy of infrequent high-dose vitamin D supplementation. In one widely reported study of 2256 community-dwelling women over 70 yr of age, an annual oral dose of 500,000 IU of vitamin D3 for 3-5 yr appeared to increase the risk of falls, particularly in the first 3 months after the vitamin D dose (267). The median baseline 25D concentration was 21.2 ng/ml (53 nmol/liter), and 1 month after treatment, the median was 48 ng/ml (120 nmol/liter).
Another study using 300,000 IU, given annually by im injection, reported no effect on falls among 9440 older individuals (268). No benefit was observed in any subgroup.
3. Meta-analyses and falls
Substantial heterogeneity among these randomized trials with regard to differences in study populations, variable treatment durations, and regimens of supplemental vitamin D and whether or not calcium was coadministered together with inconsistencies in the identification and analysis of falls among studies (e.g. falls per subject, number of falls, and number of fallers) makes collective assessment of these data difficult. Nevertheless, several meta-analyses have been published and are the subject of debate (23, 29, 30, 269-272).
Most recently, a meta-analysis reported on 26 randomized trials that enrolled 45,782 participants, mainly elderly females (23). Studies were not excluded for lack of double randomization or strict definition of falls; hence, there were a substantially larger number of included individuals than in earlier meta-analyses. Vitamin D use was associated with statistically significant reduction in the risk of falls. However, there was no difference among those receiving higher doses (>800 IU) vs. lower doses. Vitamin D appeared to be effective in both community-dwelling and institutionalized people and in those receiving vitamin D2 or D3. Perhaps reassuringly, the reduction in falls was most prominent in patients who were vitamin D deficient at baseline. Studies in which calcium was co-administered with vitamin D showed a greater effect than those where vitamin D alone was given. Falls reduction in studies without calcium coadministration did not reach statistical significance. Calcium alone was the placebo in most of the combination studies.
Therefore, despite substantial heterogeneity among studies, vitamin D supplementation is probably effective in conjunction with calcium in the prevention of falls among older individuals. The positive impact of vitamin D and calcium supplementation on fall prevention appears to be best among those who are vitamin D deficient at baseline. We recommend consideration of vitamin D plus calcium therapy in people over 65 yr of age with baseline deficiency. However, appropriate supplemental dose of vitamin D and target serum levels required to prevent falls remain hotly debated issues.
F. Muscle strength and physical performance
Many studies have examined the specific effects of vitamin D on measures of muscle function and physical performance. Comparing these studies is made very difficult by the variety of outcome measures used to assess muscle function.
1. Observational studies
A number of cross-sectional studies have reported associations between 25D levels and various parameters related to muscle function including handgrip, lower limb strength, balance, 6-min walk distance, and gait speed (273-275), although not all such studies have supported the association after multivariate adjustment (276, 277). These are summarized in Table 6.
In a population-based study of 4100 ambulatory adults aged over 60 yr, an association between serum 25D and lower-extremity function assessed by 8-ft. walk and the repeated sit-to-stand test (STST) was reported (278). The improvements were modest: 3.9% worsening in STST and 5.6% in 8-ft. walk comparing the lowest vs. the highest quintiles of 25D. Interestingly, a trend toward impaired function with longer STST was seen in patients with high normal 25D [>48 ng/ml (120 nmol/liter)] vs. lower levels [16-38 ng/ml (40-94 nmol/liter)].
Significantly poorer physical performance was seen in 1234 older individuals among those with 25D below 10 ng/ml (25 nmol/liter) and 10 -20 ng/ml (25-50 nmol/liter) vs. those higher than 30 ng/ml (75 nmol/liter) (279). In a subgroup of 979 participants followed over 3 yr, those with lower 25D levels had significantly higher risk of decline in physical performance compared with those with levels above 30 ng/ml (75 nmol/liter). Those in the intermediate 25D group of 20-30 ng/ml (50-75 nmol/liter) did not display significantly greater rates of decline.
In a prospective study of 1008 older individuals in the Netherlands, baseline 25D below 10 ng/ml (25 nmol/ liter) was associated with poorer grip strength and reduced muscle mass on DEXA after 3 yr compared with people with normal levels [>20 ng/ml (50 nmol/ liter)] (280). Those in the intermediate 25D group of 10 -20 ng/ml (25-50 nmol/liter) demonstrated significantly greater losses of muscle mass without difference in grip strength vs. those with 25D over 20 ng/ml (50 nmol/liter).
An accelerated rate of decline in physical performance over 2.5 yr on timed up and go tests (TUAG) and timed STST were seen among 769 older women with lower baseline 25D (281). However, low 25D levels are associated with frailty (273), so a range of factors not included in adjustment models may confound data interpretation.
A recent study of 714 Chinese men (>65 yr) found no association between baseline 25D levels and changes in performance measures or appendicular skeletal muscle mass over 4 yr (282). These men were not vitamin D deficient [mean baseline 25D = 31 ng/ml (78 nmol/liter)].
2. Nonrandomized studies
Few studies have examined the effect of vitamin D supplementation on muscle function in younger people. Gle-rup et al. (208) reported a case-control study in which 55 veiled Arabic women with a mean age of 32 and severe vitamin D deficiency [mean 25D = 3 ng/ml (7 nmol/liter)] were compared with 22 Danish women of similar age but higher 25D levels [19 ng/ml (47 nmol/liter)]. At baseline, all parameters of muscle function were significantly lower