Vitamin D receptor may suppress skin cancer
This is a chapter in Sunlight, Vitamin D and Skin Cancer, Second Edition, edited by Jörg reichrath.
©2013 Landes Bioscience and Springer Science+Business Media.
PDF is attached at the bottom of this page
THE VITAMIN D RECEPTOR: A Tumor Suppressor in Skin
Daniel D. Bikle
Department of Medicine andDermatology, VA Medical Center and University of California, San Francisco,
California, USA. Email: daniel.bikle@ucsf.edu
Abstract: Cutaneous malignancies including melanomas and non melanoma skin cancere (NMSC) are the most common types of cancer, occurring at a rate of over 1 million per year in the United States. The major cell in the epidermis, the keratinocyte, not only produces vitamin D but contains the enzymatic machinery to metabolize vitamin D to its active metabolite, 1,25(OH)2D, and expresses the receptor for this metabolite, the vitamin D receptor (VDR), allowing the cell to respond to the 1,25(OH)2D that it produces. In vitro, 1,25(OH)2D stimulates the differentiation and inhibits the proliferation of these cells and so would be expected to be tumor suppressive. However, epidemiologic evidence demonstrating a negative relationship between circulating levels of the substrate for CYP27B1, 25OHD, and the incidence of these malignancies is mixed, raising the question whether vitamin D is protective in the in vivo setting. UV radiation (UV), both UVB and UVA, as occurs with sunlight exposure is generally regarded as causal for these malignancies, but UVB is also required for vitamin D synthesis in the skin. This complicates conclusions reached from epidemiologic studies in that UVB is associated with higher 25OHD levels as well as increased incidence of cutaneous malignancies. Based on our own data and that reported in the literature we hypothesize that vitamin D signaling in the skin suppresses UVR induced epidermal tumor formation. In this chapter we will first discuss recent data regarding potential mechanisms by which vitamin D signaling suppresses tumor formation, then focus on three general mechanisms that mediate tumor suppression by VDR in the skin: inhibition of proliferation and stimulation of differentiation, immune regulation, and stimulation of DNA damage repair (DDR).
introduction
Over 1 million skin cancers occur annually in the United States, 80% of which are basal cell carcinomas (BCC), 16% squamous cell carcinomas (SCC), and 4% melanomas, making skin cancer by far the most common cancer afflicting humankind.1 UV radiation (UVR) from the sun is the major etiologic agent for these cancers. The highest energy UVR, UVC (below 280 nm), does not penetrate the atmosphere. Of the solar radiation that does reach the earth 95% is UVA and 5% UVB. UVB (280-320 nm), although it does not penetrate past the epidermis, is absorbed by DNA in the epidemial cells creating characteristic mutations identified as cyclobutane pyrimidine dimers (CPDs) and pyrimidine(6—4)pyrimidone photoproducts (6-4PP), which if not repaired result in C to T or CC to TT mutations, the UVB "signature" lesion2,3. UV wavelengths between 320-400 nm (UVA) are capable of penetrating into the dermis, and do their DNA damage (e.g., 8 hydroxy 2' deoxyguanosine production) primarily by oxidative processes, although at high enough dose levels UVA can produce CPDs.4 On the other hand UVB is required to convert 7-dehydrocholesterol levels in the skin to pre vitamin D3, which then isomerizes to vitamin D3. Moreover, the skin is capable of converting the vitamin D produced to its active metabolite 1,25(OH)2D,5 and this conversion is potentiated by UVR at least in part by cytokines such as TNF-a6 which are increased by UVR in the epidermis.7 Both melanocytes8 and keratinocytes9 express the vitamin D receptor (VDR) and respond to 1,25(OH)2D with reduced proliferation and increased differentiation.10,11 Sun avoidance may reduce one's risk of developing skin cancer, but this practice generally results in suboptimal levels of vitamin D in the body. In an analysis by Lucas et al.,12 the global disease burden due to UVR is substantially less than the disease burden due to vitamin D deficiency. Vitamin D supplementation can compensate, but the skin remains the major site of vitamin D availability for most of the world's population. Moreover, low dose UVR may be protective against skin cancer via the vitamin D signaling mechanisms that will be reviewed in this article, and some epidemiologic evidence is consistent with a potential benefit of low dose UVR. For example, in the study by Armstrong and Kricker,13 a slight decrease in the incidence of SCC, BCC, and melanomas in 10 US populations was observed when the solar UV measurement was increased from 100 to 110, although higher levels increased the incidence. This same group,14 evaluating data from the Australian population, did not find a significant increase in SCC with time spent out of doors in the general population. Rosso et al.15 in a multicenter European study did not find a significant increase in SCC below a threshold of 70,000 accumulated hours of sunshine, although the development of BCC had a lower threshold. In this chapter, after a review of vitamin D metabolism and VDR function, we will examine potential mechanisms that have been proposed for vitamin D induced antitumor mechanisms in general, then focus on those mechanisms that have been shown to be operative in the epidermis.
vitamin d metabolism
Vitamin D3 is produced from 7-dehydrocholesterol (7-DHC). Irradiation of 7-DHC with UVB produces pre vitamin D3, which subsequently undergoes a temperature-dependent rearrangement of the triene structure to form vitamin D3, lumisterol, and tachysterol. This process is relatively rapid and reaches a maximum within hours16-18 although both the degree of epidemial pigmentation and the intensity of exposure influence the time required to achieve this maximal concentration of pre vitamin D3. With continued UV exposure the biologically inactive lumisterol and tachysterol accumulate eliminating the risk of excessive production of vitamin D. Sunlight exposure increases melanin production, which can absorb UVB, and so provides another mechanism by which excess vitamin D3 production can be prevented. The intensity of UVR is dependent on latitude and season. In Edmonton, Canada (52°N) very little vitamin D3 is produced in exposed skin from mid-October to mid-April, while in San Juan (18°N) the skin is able to produce vitamin D3 all year long.19 Clothing and sunscreen effectively prevent vitamin D3 production in the covered areas. Vitamin D3 produced in the skin can be carried to the liver and other tissues for further metabolism to 25-hydroxyvitamin D (25OHD) and then to the kidney to produce 1,25(OH)2D by the enzyme CYP27B1. However, as noted above, the keratinocyte contains the entire pathway for 1,25(OH)2D production from vitamin D.
The production of 1,25(OH)2D in the skin is under quite different regulation compared with its production by the kidney. In the kidney parathyroid hormone (PTH), fibroblast growth factor 23 (FGF23) and 1,25(OH)2D itself are the principal hormonal regulators: PTH stimulates, whereas FGF23 and 1,25(OH D inhibit 1,25(OH)2D production. Keratinocytes respond to PTH with increased 1,25(OH)2D production, but these cells do not have the classic PTH receptor and do not respond to cyclic AMP5 unlike the kidney. The effect of FGF23 on keratinocyte CYP27B1 expression or function has not been reported. Furthermore, unlike the kidney, 1,25(OH)2D does not directly affect CYP27B1 expression in keratinocytes. Rather, 1,25(OH)2D regulates its own levels in the keratinocyte by inducing CYP24, the catabolic enzyme for 1,25(OH)2D3.20 In the keratinocyte the major regulators of1,25(OH)2D production are cytokines such as tumor necrosis factor-a (TNF)6 and interferon-g (IFN).21 These cytokines are activated in the skin by UVB, which of course also increases the substrate via increased vitamin D production.
VITAMIN D RECEpTOR: MECHANISM OF ACTION
The VDR is a member of the nuclear hormone receptor superfamily.22 These members are characterized by a highly conserved DNA binding domain characterized by two zinc fingers and a structurally conserved ligand binding domain that has at its C-terminal end the AF2 domain to which coactivator complexes bind.23 The ligand binding domain also serves as the region to which VDR binds to its transcriptional partners, RXR being the major one. In general ligand (ie. 1,25(OH)2D) binding is required for the VDR/RXR heterodimer to form and bind to those regions on the DNA called vitamin D response elements (VDRE). Ligand binding also alters the structure ofthe VDR with major movement of helix 12 (C-terminus) into position to enclose the ligand while exposing sites on the VDR in helices 3, 5 and 12 to which coactivators bind. These coactivators can in turn recruit chromatin modifying enzymes such as histone acetyl transferases (SRC, CBP/ p300, pCAF) and DNA demethylases or proteins that bridge the gap between the VDRE and the transcription machinery (Mediator complex) including TATA associated factors, TFIIb, and RNA polymerases (primarily RNA pol II). In the absence of ligand binding sites for corepressors are exposed. These corepressors recruit another set of chromatin modifying enzymes such as histone deacetylases and DNA methyl transferases.24 The most common VDRE is comprised of two head to tail half sites of hexanucleotides separated by 3 nucleotides, referred to as DR3 VDREs. The sequence of these DR3 half sites is heterogeneous, with a consensus approximated by RGKTSA where R = A or G, K = G or T, S = C or G. Moreover, many VDREs are not DR3s, although DR3s tend to have the highest affinity for VDR/RXR heterodimers.
The VDREs can be quite distant from the transcription start site of the gene being regulated, occurring in introns, between genes, and in either a 5' or 3' relationship to the coding region.25 Moreover, putative VDREs as demonstrated by techniques such as ChIP-seq, in which binding sites to the genome by VDR are identified using a combination of chromatin immunoprecipitation of the VDR to DNA followed by high throughput sequencing of those binding regions, number in the thousands, with a substantial degree of cell/tissue specificity.26 Most genes have several VDREs. In a review of two such ChIP-seq studies Carlberg et al.26 noted that the study in a lymphoblastoid line identified 2776 VDREs for 232 genes whereas the study in THP-1 monocytes identified 1820 VDREs for 638 genes. In the latter study 408 of the genes were upregulated, 230 downregulated. Most of the VDREs for the upregulated genes were within 400kbp ofthe transcription start site; this was less true for the downregulated genes. Only 93 of the upregulated genes had VDREs within 30kbp of the transcription start site. Moreover, only 31.7% of the VDREs were DR3s. These two studies had a mere 18% overlap of the VDREs identified, but differed not only in cell line but dose and time after 1,25(OH)2D was administered before the cells were analyzed. Earlier microarray studies had likewise demonstrated the many genes regulated by 1,25(OH)2D, and the surprising lack of consensus from one study to the next perhaps due to tissue specificity and/or differences in dose and time of 1,25(OH)2D exposure. These studies demonstrate the diversity of vitamin D regulated genes and diversity in type and location of VDREs. Such studies have revolutionized our concepts of the scope and means of vitamin D signaling, and reveal many potential mechanisms by which vitamin D signaling can regulate cancer formation. In this regard an unbiased systems biology approach mapping genetic loci underlying susceptibility to skin cancer put the VDR in the center of a complex set of networks linking regulation of barrier function, inflammation, and tumor formation.27
The VDR is essential for nearly all actions of 1,25(OH)2D and its analogs. Tumors that are unresponsive to vitamin D have either lost their ability to produce 1,25(OH)2D (ie. decreased CYP27B1),28,29 increased their metabolism of1,25(OH)2D via upregulation of CYP24A1,30 lost VDR transcriptional activity through post translational alterations in RXR,31 or decreased their VDR expression. The latter may be secondary to increased activity in tumors of inhibitors of VDR expression such as SNAIL32 and SLUG,33 increased methylation of the VDR promoter,34 or increased expression of miRNA125b, an inhibitor of VDR expression.35 In melanoma cell lines the administration of 5-aza cytidine (to inhibit DNA methyltransferase) and trichostatin (to inhibit HDAC activity) could restore responsiveness to 1,25(OH)2D by increasing VDR levels and reducing miR125b expression.36
MECHANISMS Of TUMOR SUppRESSION By VITAMIN D: GENERAL
The demonstration that many genes and pathways are influenced by vitamin D signaling has opened up a large number of potential means by which vitamin D signaling can control tumor growth (recent reviews in refs. 37,38) (Fig. 1).
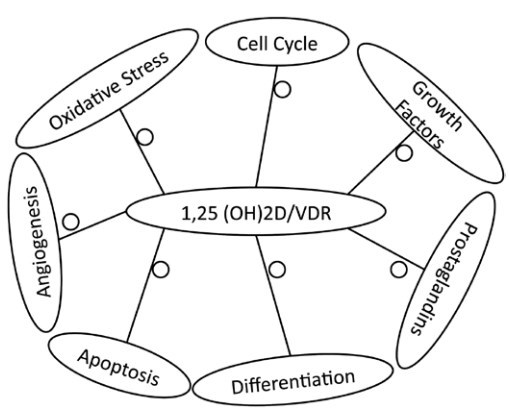
Figure 1.
Multiple mechanisms by which 1,25(OH)2D/VDR suppress tumor formation. Vitamin D signaling has the potential to suppress tumor formation by affecting a number of pathways. Angiogenesis is suppressed by reducing the expression of VEGF and its receptors. Oxidative stress is reduced by increasing the expression of enzymes that reduce ROS. The cell cycle is inhibited by increasing the expression of cell cycle inhibitors and decreasing the expression of cyclins and cyclin dependent kinases. The expression and/or activation of growth factors is inhibited (for growth factors that promote proliferation) or stimulated (for growth factors that inhibit proliferation). Prostaglandin synthesis is inhibited, the expression of their receptors is reduced, and the expression of enzymes that degrade prostaglandins in increased. On the other hand differentiation of the cells is increased, and apoptosis of damaged cells is stimulated.
Cell Cycle Regulation
Regulationby 1,25(OH)2D of the cell cycle in a number of cells, normal and malignant, has been demonstrated. This results from an upregulation of cell cycle inhibitors such as p21cip and p27kip (cyclin-dependent kinase inhibitors)39 and retinoblastoma like protein 2 and retinoblastoma binding protein 640 and decreased expression of cyclins41 and cyclin dependent kinases.42 In addition 1,25(OH)2D increases the interaction of FoxO proteins (tumor suppressors controlling proliferation43) with VDR and FoxO regulators Sirt1 and protein phosphatase 1 that maintain FoxO in the nucleus by blocking MAPK phosphorylation.44 Increased c-MYC expression and activity are frequently found in cancer.45 c-MYC induces the expression of a number of cell cycle regulatory genes such as cyclin D2 and cdk4. 1,25(OH)2D inhibits the expression of c-MYC,46 and c-MYC expression is increased in the skin and gut of VDR null mice.47
Growth Factors
1,25(OH)2D and its analogs regulate a number of growth factor pathways. Insulin like growth factor (IGF) stimulated proliferation ofbreast and prostate cells is reduced by 1,25(OH)2D via its induction of IGF-I binding protein 3.48,49 TGFP2 exerts antiproliferative actions in epithelial cells. 1,25(OH)2D and its analogs increase the expression of TGFp2 and TGFp receptors in breast and prostate cancer cells,40,42,50 while suppressing the expression of the latent TGFp-binding protein.42,51 GDF15 (growth differentiation factor 15) is a member of the TGFp superfamily, and like TGFp is antiproliferative in prostate cancer cells. Its expression is increased by 1,25(OH)2D.52,53 Bone morphogenic proteins (BMPs) are also members of the TGFb superfamily that have been found to be dysregulated in certain cancers.54 The expression of several BMPs is regulated by 1,25(OH)2D and its analogs in a number of malignant cell lines.41,42,55 Wnt/p-catenin signaling will be dealt with in depth when we focus on vitamin D regulated pathways in the skin, but this pathway has been extensively studied in the colon based on the frequency of mutations in the adenomatous polyposis coli (APC) gene in colon cancer.56 In the canonical pathway of wnt/p-catenin signaling the APC complex that would otherwise bind and phosphorylate p-catenin, targeting it for proteosomal degradation, is inactivated, allowing p-catenin to move to the nucleus where it binds to LEF/TCF leading to transcription of genes involved with proliferation. 1,25(OH)2D/VDR binds to p-catenin, preventing its movement into the nucleus and/or binding to LEF/TCF.57,58 Moreover, by increasing the levels of E-cadherin, which binds p-catenin in the plasma membrane, 1,25(OH)2D can further reduce the translocation of p-catenin into the nucleus.57,58 Furthermore, 1,25(OH)2D can suppress wnt signaling by stimulating the expression of the wnt antagonist DKK-1.59 Cystatin D, an inhibitor of several cysteine proteases of the cathepsin family that appear to be involved in wnt signaling, has likewise been shown to be a target gene of 1,25(OH)2D.60 The induction of cystatin D and other 1,25(OH)2D target genes such as E-cadherin appear to involve a non genomic action requiring calcium activation of RhoA-ROCK-p38MAPK-MSK in colon cancer cells.61 We have shown that this pathway requires the 1,25(OH)2D induced calcium receptor in keratinocytes.62 These and other studies point to the interaction between calcium and vitamin D signaling in the regulation of tumor formation,63 an interaction that to date has received little attention.
Apoptosis
In addition to inhibiting proliferation, 1,25(OH)2D promotes apoptosis in a number of malignant cell lines in part by downregulation of anti-apoptotic genes Bcl-2 and Bcl-XL64,65 and upregulation of the proapoptotic gene GOS2 (G0G1 switch gene 2).41,66 Transcripts of other pro-apoptotic genes increased by 1,25(OH)2D include death-associated protein-3, caspase 8 apoptosis-related cysteine peptidase, and fas-associated death domain-like apoptosis regulator as well as a number of caspases.40 Telomerase is a mechanism that enables cancer cells to escape apoptosis. 1,25(OH)2D suppresses telomerase expression by inducing miRNA498, a transcript in the complementary strand of CTC-360P6.67 Of interest is this miRNA has its own VDRE.67
Oxidative Stress
As noted previously inthe discussion ofUVA induced effects onthe epidermis, oxidative stress can lead to oxidative DNA damage, marked by 8 hydroxy 2'-deoxyguanosine. In VDR knockout mice, 8 hydroxy 2'-deoxyguanosine levels are increased in the colon68 and reduced by vitamin D supplementation in humans.69 1,25(OH)2D induces several antioxidant enzymes in cancer cells including thioredoxin reductase 1,40,42 superoxide dismutase,42,52 and glucose-6 phosphate dehydrogenase.51 The induction of genes associated with DNA repair will be discussed at greater length when we focus on UVB damage to the epidermis, but the induction by 1,25(OH)2D of GADD45a (growth arrest and DNA-damage inducible a), p53, RAD23B, PCNA, and DAP-1a may all contribute to this aspect of tumor suppression by 1,25(OH)2D/VDR.40,66,70
Prostaglandins
Prostaglandins have been shown to stimulate cancer cell growth.71 1,25(OH)2D blocks prostaglandin signaling by inhibiting COX2 expression and that of prostaglandin receptors while increasing the expression of hydroxyprostaglandin dehydrogenase 15-NAD, the prostaglandin inactivating enzyme.53,72
Angiogenesis
Growing tumors require a blood supply. 1,25(OH)2D inhibits angiogenesis by blocking the expression and function of VEGF (vascular endothelial growth factor),73-75 and mice lacking VDR had larger and more vascular tumors when implanted with prostate cells from TRAMP mice.76
Immune System
The immune system plays an important protective role in cancer protection77 as evidenced by the increased numbers of malignancies in immunosuppressed hosts including SCCs in immunosuppressed renal transplant patients.78 This mechanism will be dealt with in more depth when we focus on the skin, as it has not received much study in the context of tumor protection in general by 1,25(OH)2D. In fact by stimulating innate immunity, which would promote inflammation, and suppressing adaptive immunity, which would blunt immune surveillance one might expect 1,25(OH2D to promote rather thanblock tumor development via its actions on the immune system. As such, this mechanism of action of 1,25(OH)2D and its receptor with respect to cancer development requires further study.
MECHANISMS OF TUMOR SUppRESSION By 1,25(OH)2D/VDR IN THE EpiDERMIS
The potential for vitamin D signaling as protection against epidemial tumor formation was demonstrated when Zinser et al.79 demonstrated that 85% of the VDR null mice but none of the controls developed skin tumors within two months following 7,12 dimethylbenzanthracene (DMBA) administration. These were primarily papillomas. These results have been confirmed using topical administration of DMBA/TPA.80 However, although only papillomas were seen in the VDR null mice, RXRa null mice developed both BCC and SCC.80 Subsequently, Ellison et al.81 and our own group82demonstrated that VDR null mice were also more susceptible to tumor formation following UVB, and many of the tumors were SCC and BCC. The appearance of BCC in these studies is surprising since the typical malignancy induced in mouse skin by UVR, ionizing radiation, or chemical carcinogens is SCC not BCC.83 Given that BCC generally result from increased hedgehog (Hh) signaling,84 and that lack of VDR results in BCC when p-catenin signaling is increased,85 we became interested in the relationship between vitamin D, Hh, and p-catenin signaling in tumor suppression. The lack of a normal innate immune response in CYP27B1 null mice to wounding86 or infection87 and the increased numbers of SCC in immunocompromised patients78 suggested that disruption of the immune system might contribute to the increased susceptibility to tumor formation when vitamin D signaling was impaired. Moreover, we82 noted a reduction in clearance in CPDs following UVB exposure ofthe skin of VDR null mice, suggesting that disruption ofDNA damage repair was playing a role in tumor susceptibility in these mice. In what follows I will examine three potential mechanisms and pathways within those mechanisms for their contribution to the role of VDR as a tumor suppressor: regulation of proliferation and differentiation with particular attention to the hedgehog (Hh) and wnt/p-catenin pathways, immunoregulation, and DNA damage repair.
Vitamin D Regulation of Epidermal proliferation and Differentiation
The epidermis is composed of four layers of keratinocytes at different stages of differentiation (reviewed in ref. 10). The basal layer (stratum basale, SB) rests on the basal lamina separating the dermis and epidermis. Within this layer are the stem cells. These cells proliferate, providing the cells for the upper differentiating layers. The basal cells are characterized by keratins K5 and K14 as well as the stem cell marker K15 and integrin a6p4. As the cells migrate upward from this basal layer into the spinous layer (stratum spinosum, SS) they initiate the production of the keratins K1 and K10, the keratins characteristic of the more differentiated layers of the epidermis. Cornified envelope precursors such as involucrin also appear in the spinous layer as does the enzyme transglutaminase K, responsible for the e-(g-glutamyl)lysine cross-linking of these substrates into the insoluble cornified envelope. Migrating further into the granular layer (stratum granulosum,{SG}), lying above the spinous layer, the cells acquire the electron-dense keratohyalin granules containing profilaggrin and loricrin that give the SG its name. Loricrin is a major component of the cornified envelope. Filaggrin serves to bundle the keratin filaments, but also when proteolyzed is thought to contribute to the hydration of the outer layers. The granular layer also contains lamellar bodies-lipid, enzyme, and antimicrobial peptide filled structures that fuse with the plasma membrane, divesting their contents into the extracellular space between the SG and stratum corneum (SC). The secreted enzymes process the lipids that contribute to the permeability barrier of the epidermis in conjunction with the keratin bundles and cornified envelope. The antimicrobial peptides provide a barrier against infectious organisms in the SC.
1,25(OH)2D increases essentially every step of this differentiation process88-93 while inhibiting proliferation at least at concentrations above 1nM. These actions complement those of calcium,62 the response to which is enhanced by 1,25(OH)2D via its induction of the calcium receptor,94,95 and the phospholipase C enzymes96-98 that regulate intracellular calcium and other signaling molecules critical for the differentiation process. The antiproliferative effects are accompanied by a reduction in the expression of c-myc99 and cyclin D1100 and an increase in the cell cycle inhibitors p21cip and p27kip. In addition, 1,25(OH)2D and its receptor regulate the processing of the long chain glycosylceramides that are critical for permeability barrier formation101 and induce the receptors, toll like receptor 2 (TLR2) and its coreceptor CD14, that initiate the innate immune response in skin.86 Activation of these receptors leads to the induction of CYP27B1 (the enzyme that produces 1,25(OH)2D), which in turn induces cathelicidin resulting in the killing of invasive organisms.86,102 Deletion of either VDR103,104 or CYP27B1105 results in defects in the differentiation process leading to an abnormal barrier and increased proliferation of the epidermis with a defective innate immune response.86 Two pathways that appear to be important in vitamin D signaling in the epidermis with respect to proliferation and differentiation that we believe underlie the predisposition of the VDR null mouse to tumor formation are the Hh and wnt/p-catenin pathways.
The Hedgehog (Hh) Pathway (Fig. 2).
In the skin sonic hedgehog (Shh) is the ligand for patched (Ptch) 1, a 12 transmembrane domain protein that in the absence of Shh inhibits the function of another membrane protein smoothened (Smo). Smo in turn maintains a family of transcription factors, Gli1 and Gli2 in particular, in the cytoplasm bound to Suppressor of fused (Sufu).106,107 When Shh binds to Ptch 1, the inhibition of Smo is relaxed, Gli1 and 2 are released from Sufu and move into the nucleus where they initiate transcription of a number of factors including each other as well as Ptch 1, the anti apoptotic factor bcl2, cyclins D1 and D2, E2F1, cdc45 (all of which promote proliferation), while suppressing genes associated with keratinocyte differentiation such as K1, K10, involucrin, loricrin and the VDR.108-112
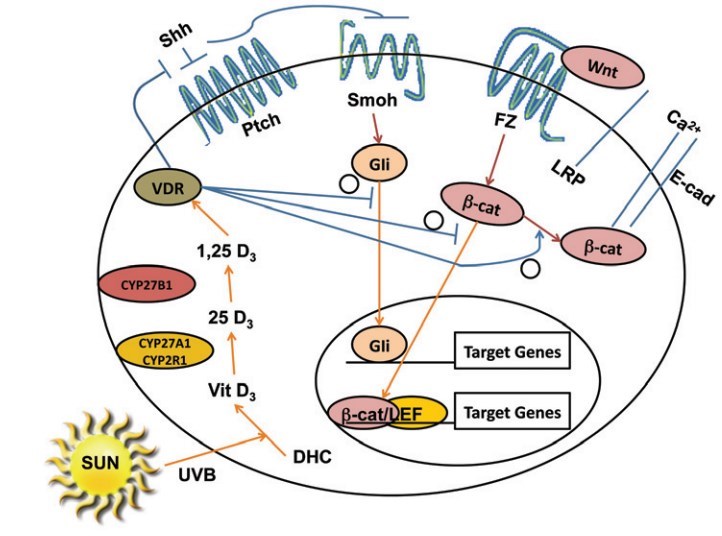
Figure 2. Regulation of Hh and wnt/p-catenin signaling by 1,25(OH)2D/VDR. The keratinocyte expresses VDR and is capable of making its own 1,25(OH)2D3 from the vitamin D3 produced from 7-dehydrocholesterol (DHC) under the influence of UVB, as it has both CYP27A1/CYP2R1 (which convert vitamin D3 to 25OHD3) and CYP27B1 (which converts 25OHD3 to 1,25(OH)2D3). 1,25(OH)2D/VDR suppresses Shh and gli 1 expression, inhibiting the Hh pathway in keratinocytes. 1,25(OH)2D/VDR binds p-catenin and induces E-cadherin expression reducing the amount of p-catenin available for binding to TCF/LEF in the nucleus limiting its transcriptional activity. In combination these actions reduce the proliferative actions of Shh and p-catenin signaling in keratinocytes, limiting their ability to induce tumors in the skin.
The appearance of BCC is characteristic of tumors formed when Hh signaling is disrupted,113 although activation of Hh signaling also predisposes to UVR induced SCC formation.114 VDR null animals overexpress elements of the Hh signaling pathway in their epidermis and the epidemial portion (utricles) of the hair follicles.82 Moreover, 1,25(OH)2D suppresses the expression of all elements of the Hh pathway in a dose dependent fashion that requires the VDR82,115 and reduces tumor growth in Ptch 1null mice. The promoters of Shh and Gli1 have binding sites for VDR116 suggesting that the effects of 1,25(OH)2D on these genes is direct. However, vitamin D has also been shown to bind to and inhibit the actions of smoothened (Smo) directly without seeming to require further metabolism to 1,25(OH)2D.117,118
The wnt/pp-Catenin Pathway (Fig. 2). Wnt signaling via activation of p-catenin has a complex role in VDR function as discussed briefly earlier. In the canonical pathway the receptor for wnt ligands is a family of seven-transmembrane Frizzled receptors and an LRP5 or LRP6 co-receptor. When wnt binds to this complex disheveled (Dvl) is phosphorylated resulting in disruption of the axin/APC complex and inhibition of glycogen synthase kinase 3p GSK-3p. In the basal state GSK-3p phosphorylates the serine(s) within exon 3 of p-catenin resulting in its degradation by the E3 ubiquitin ligase. Wnt signaling, by blocking this phosphorylation, increases the availability of p-catenin in the nucleus, where it binds to transcription factors of the T-cell factor (TCF) and lymphoid enhancer factor (LEF) families to promote expression of genes such as cyclin D1 and c-myc119 important for proliferation. p-catenin also forms part of the adherens junction complex with E-cadherin where it may play an important role in keratinocyte differentiation.120 Tyrosine phosphorylation of E-cadherin, as occurs after calcium administration to keratinocytes, promotes the binding of p-catenin and other catenins to the adherens junction complex120,121 making it less available for transcriptional activity. 1,25(OH)2D increases E-cadherin expression.122 Overexpression and/or activating mutations in the p-catenin pathway lead to skin tumors, in this case pilomatricomas or trichofolliculomas (hair follicle tumors).123-125 As noted earlier VDR binds to p-catenin, and reduces the transcriptional activity of p-catenin in a 1,25(OH)2D dependent fashion.57 On the other hand binding of p-catenin to VDR in its AF-2 domain enhances the 1,25(OH)2D dependent transcriptional activity of VDR.58 Palmer et al.85 evaluated the interaction between VDR and p-catenin in transcriptional regulation in keratinocytes, and identified putative response elements for VDR and p-catenin/LEF in a number of genes. These interactions were either positive or negative, depending on the gene being evaluated. The hypothesis put forward is that genes in which the interaction was positive (ie. stimulated transcription) benefited from p-catenin acting as a coactivator for VDR on VDREs, whereas in situations where the interaction was negative (ie. suppression of transcription) VDR prevented p-catenin from binding to TCF/LEF required for transcription in those genes. We100 have found in keratinocytes that knockdown of VDR reduces E-cadherin expression and formation of the p-catenin/E-cadherin membrane complex resulting in increased p-catenin transcriptional activity, whereas 1,25(OH)2D administration has the opposite effect. This was associated with increased (with VDR knockdown) or decreased (with 1,25(OH)2D administration) keratinocyte proliferation and cyclin D1 expression. On the other hand Cianferotti et al.126 found a reduction in proliferation of keratinocytes in the dermal portion of the hair follicle (below the bulge) in VDR null mice, and no stimulation of proliferation when p-catenin was overexpressed in these cells in contrast to the stimulation of proliferation in control animals. Thus VDR/p-catenin interactions can be positive or negative, depending on the gene/cell/function being evaluated, but in the epidermis in the absence of VDR, the unchecked activity of p-catenin appears to be proliferative and inhibitory of differentiation resulting in BCC.
Vitamin D Regulation of Immune Function in the Skin
VDR and CYP27B1 are found in professional immune cells, namely dendritic cells, macrophages, and lymphocytes127,128 responsible for both innate and adaptive immune responses as well as in epithelial cells expressing the components of the innate immune response. 1,25(OH)2D regulates the proliferation and function129 of these cells. Although it is not clear the extent to which dysregulated immune function contributes to cancer development in the skin, a link between inflammation and cancer susceptibility in the skin involving VDR has been established.27
Adaptive Immunity. The adaptive immune response involves the ability of T and B lymphocytes to produce cytokines and immunoglobulins, respectively, in response to antigens presented to them by cells such as macrophages and dendritic cells. 1,25(OH)2D suppresses the adaptive immune response by inhibiting proliferation, immunoglobulin production, and differentiation of B-cell precursors into plasma cells.128 1,25(OH)2D inhibits T cell proliferation130 and the differentiation of CD4 cells into Th1 cells capable of producing IFN-g and IL-2 and activating macrophages131 and Th17 cells capable of producing IL17 and IL22.132,133 On the other hand 1,25(OH)2D stimulates IL-4, IL-5, and IL10 production134 by increasing CD4 cell differentiation into Th2 and regulatory T cells (Treg).135 The IL-10 so produced is one means by which Treg block Th1 and Th17 development. Part of these effects is mediated by the negative impact of 1,25(OH)2D on the maturation and antigen presenting capability of dendritic cells.136 It is unclear if this suppression of the adaptive immune system alters tumor surveillance in the skin.
Innate Immunity . The innate immune response involves the activation of toll-like receptors (TLRs)137 that serve as transmembrane pathogen-recognition receptors detecting specific membrane patterns (PAMP) shed by a wide variety of infectious agents.138 Activation of TLRs leads to the induction of antimicrobial peptides and reactive oxygen species, which kill the organism. Cathelicidin is the best studied of these antimicrobial peptides. The expression of cathelicidin is induced by 1,25(OH)2D in both myeloid and epithelial cells,139,140 cells that also express CYP27B1 and so are capable of producing 1,25(OH)2D needed for this induction. Stimulation of TLR2 in macrophages141 or keratinocytes86 results in increased CYP27B1 expression, which in the presence of adequate substrate (25OHD) induces cathelicidin expression. Lack of substrate (25OHD), VDR, or CYP27B1 blunts the ability of these cells to respond with respect to cathelicidin production.86,140,141
The major cells involved in adaptive immunity in the skin include the Langerhans cells, dendritic cells, and T cells. The Langerhans cells are dendritic like cells within the epidermis that when activated by invading organisms migrate to the lymph nodes serving the skin where they present the antigens to the T cells, initiating the adaptive immune response.142 Keratinocytes, on the other hand, are equipped with toll like receptors that enable them to respond to invading organisms with elaboration of antimicrobial peptides such as cathelicidin.102 However, cathelicidin also induces an inflammatory response.143 UVB leads to a reduction in Langerhans cells and blunts their antigen presenting activity,144-146 but stimulates the innate immune function of keratinocytes perhaps as a consequence of UVB induced vitamin D/1,25(OH)2D production in the skin.147,148
The potential role of altered skin immunity by UVB with respect to skin carcinogenesis was suggested by Kripke and Fisher.149 They found that skin tumors originally induced in mice by chronic UVR, would grow when transplanted into mice that had been UV irradiated but not when transplanted into control mice. The role of 1,25(OH)2D production in UVR immunosuppression is not clear. Topical application of high concentrations of 1,25(OH)2D protected against UVR induced suppression of contact hypersensitivity in the mouse,150 but a study in humans by the same group showed suppression of delayed hypersensitivity (Mantoux test) by topical 1,25(OH)2D.151 These data are limited, but raise some concern about the balance between innate and adaptive immunity in tumor surveillance, and how that balance is affected by vitamin D.
Vitamin D Regulation of the DNA Damage Response
DNA damage repair (DDR) is the means by which UVR and chemical induced DNA damage is prevented from producing fixed DNA mutations.152 DDR involves a cascade of damage recognition, repair and signal transduction that coordinates the response of the cell to DNA damage. DDR activates checkpoints that delay the cell cycle, provides time for repair, and directs damaged cells into senescent or apoptotic pathways. DDR involves a number of components, is well orchestrated, tightly controlled, and highly accurate in normal primary cells such that the spontaneous mutation rate is very low, and changes in copy number are negligible.153-155 With malignant transformation DDR becomes less controlled, and mutation rates and copy number abnormalities increase by orders of magnitude.153,154,156,157 Nucleotide excision repair (NER) is the principal means by which UVR damage is repaired, enabling repair before DNA replication begins. This is important as NER plays a major role in reducing the amount of damage that becomes fixed as mutations during replication.158-160 During NER, the DNA damage is recognized, the DNA unwound around the lesion, and 30 base pair portions of DNA containing the lesion are excised by endonucleases such as XPF and XPG followed by fill in with DNA polymerases such as Pol S,e,K.
The NER process has two main branches involving different mechanisms for the initial recognition of DNA damage161: transcription coupled repair (TCR) during which DNA polymerases stop replication at the site of the lesion until it is repaired,162-166 and global genomic repair (GGR), during which non-transcribed regions of the genome are repaired.167 Keratinocytes in the epidermis of mice lacking VDR are deficient in DDR as demonstrated by a reduced rate of clearing CPDs and 6,4PPs following UVB.168 Moreover, 1,25(OH)2D increases CPD clearance in VDR intact mice.169,170 These actions have been demonstrated with 1,25(OH)2D analogs that are not thought to have genomic activity.169 However, at least part of this enhancement of CPD clearance is due to the upregulation of two genes important for DDR: XPC (xeroderma pigmentosum complementation group C) and DDB2 (damage-specific DNA binding protein 2 also known as XPE).168,171 Furthermore, 1,25(OH)2D has been shown to increase the levels of p53, which could enhance apoptosis in those cells bearing excess DNA damage,170 and reduce UVR induced oxidative stress contributing to the DNA damage.170 As such these actions of vitamin D signaling on DDR contribute to the reduced susceptibility of normal skin to UVB induced tumor formation.
Conclusion
The VDR is present in nearly every cell in the body . Moreover, the enzyme, CYP27B1, required for the production of the VDR ligand, 1,25(OH)2D, is likewise widely distributed. Because of its abundance of 7-DHC, the epidermis is unique in its capability to produce vitamin D, metabolize it to 1,25(OH)2D, and respond to 1,25(OH)2D in a number of ways. Recent data from microarray and ChIP-seq studies have demonstrated hundreds, perhaps thousands of genes regulated by 1,25(OH)2D/VDR via VDREs which are located throughout the gene. The selection of the genes regulated by 1,25(OH)2D/VDR at any one time is cell specific and most likely dose and time specific with respect to exposure to 1,25(OH)2D. As a result of these studies, numerous pathways have been discovered by which 1,25(OH)2D/VDR may prevent cancer. In the skin UVB is critical for vitamin D production, but UVB is also the major cause of skin cancer. This chapter examines the question of whether the beneficial effects of UVB on vitamin D production can counter the harmful effects on carcinogenesis. Epidemiologic data suggest that there may be a threshold below which UVR is not carcinogenic, a threshold that would suffice for adequate vitamin D production. Conceivably, vitamin D production at such levels of UVB exposure might even be protective. Three potential mechanisms for such protection were examined.
The first mechanism focuses on the role of vitamin D signaling in keratinocyte proliferation and differentiation. In particular, two pathways affecting proliferation and differentiation, namely the hedgehog and wnt/p-catenin pathways, were evaluated. Mice lacking the VDR have increased expression of the hedgehog pathway and increased activation of the wnt/p-catenin pathway. 1,25(OH)2D suppresses both pathways in cells containing the VDR. Overexpression of the hedgehog and wnt/p-catenin pathways lead to increased proliferation and decreased differentiation associated with tumor development.
The second mechanism involves the role of vitamin D signaling in the immune system of the skin. The role of the immune system in epidermal carcinogenesis is not clear. However, in an unbiased genomic examination of pathways associated with tumor susceptibility, inflammation, keratinocyte differentiation, and tumor formation were linked through the VDR. 1,25(OH)2D/VDR promotes innate immunity but suppresses adaptive immunity. Whether this is beneficial regarding tumor development requires further study.
The third mechanism is DNA damage repair (DDR). The epidermis of VDR null mice show impaired DDR following UVR. 1,25(OH)2D accelerates DDR by what appear to be genomic and non genomic actions. On teleologic grounds one might anticipate that the skin has developed mechanisms to protect itself from the harmful effects of UVR. Vitamin D production, metabolism, and regulation of the processes described in this chapter may play a key role in this protection.
Acknowledgments
I would like to acknowledge the collaborators whose work I have referenced, namely Drs. Zhongjian Xie, Chialing Tu, Arnaud Teichert, Kumar Pillai, Yuko Oda, Yan Jiang, Dennis Oh, James Cleaver; the administrative assistance of Ms Teresa Tong and Victoria Lee; and the funding by a VA Merit Review, NIH RO1 AR050023, PO1 AR39448, and DOD CA110338
References
1. Greenlee RT, Hill-Harmon MB, Murray T, Thun M. Cancer statistics, 2001. CA Cancer J Clin 2001; 51:15-36; PMID:11577478; http://dx.doi.org/10.3322/canjclin.51.1.15.
2. Freeman SE, Hacham H, Gange RW, Maytum DJ, Sutherland JC, Sutherland BM. Wavelength dependence of pyrimidine dimer formation in DNA of human skin irradiated in situ with ultraviolet light. Proc Natl Acad Sci U S A 1989; 86:5605-9; PMID:2748607; http://dx.doi.org/10.1073/pnas.86.14.5605.
3. Hussein MR. Ultraviolet radiation and skin cancer: molecular mechanisms. J Cutan Pathol 2005; 32:191-205; PMID:15701081; http://dx.doi.org/10.1111/j.0303-6987.2005.00281.x.
4. Besaratinia A, Synold TW, Chen HH, Chang C, Xi B, Riggs AD, et al. DNA lesions induced by UV A1 and B radiation in human cells: comparative analyses in the overall genome and in the p53 tumor suppressor gene. Proc Natl Acad Sci U S A 2005; 102:10058-63; PMID:16009942; http://dx.doi.org/10.1073/ pnas.0502311102.
5. Bikle DD, Nemanic MK, Gee E, Elias P. 1,25-Dihydroxyvitamin D3 production by human keratinocytes. Kinetics and regulation. J Clin Invest 1986; 78:557-66; PMID:2426308; http://dx.doi.org/10.1172/JCI112609.
6. Bikle DD, Pillai S, Gee E, Hincenbergs M. Tumor necrosis factor-alpha regulation of 1,25-dihydroxyvitamin D production by human keratinocytes. Endocrinology 1991; 129:33-8; PMID:1675987; http://dx.doi. org/10.1210/endo-129-1-33.
7. Muthusamy V, Piva TJ. The UV response of the skin: a review of the MAPK, NFkappaB and TNFalpha signal transduction pathways. Arch Dermatol Res 2010; 302:5-17; PMID:19756672; http://dx.doi. org/10.1007/s00403-009-0994-y.
8. Colston K, Colston MJ, Fieldsteel AH, Feldman D. 1,25-dihydroxyvitamin D3 receptors in human epithelial cancer cell lines. Cancer Res 1982; 42:856-9; PMID:6895862.
9. Pillai S, Bikle DD, Elias PM. 1,25-Dihydroxyvitamin D production and receptor binding in human keratinocytes varies with differentiation. J Biol Chem 1988; 263:5390-5; PMID:2451669.
10. Bikle DD. Vitamin D and the skin: Physiology and pathophysiology. Rev Endocr Metab Disord 2012; 13:3-19; PMID:21845365; http://dx.doi.org/10.1007/s11154-011-9194-0.
11. Colston K, Colston MJ, Feldman D. 1,25-dihydroxyvitamin D3 and malignant melanoma: the presence of receptors and inhibition of cell growth in culture. Endocrinology 1981; 108:1083-6; PMID:6257495; http://dx.doi.org/10.1210/endo-108-3-1083.
12. Lucas RM, McMichael AJ, Armstrong BK, Smith WT. Estimating the global disease burden due to ultraviolet radiation exposure. Int J Epidemiol 2008; 37:654-67; PMID:18276627; http://dx.doi.org/10.1093/ije/ dyn017.
13. Armstrong BK, Kricker A. The epidemiology of UV induced skin cancer. J Photochem Photobiol B 2001; 63:8-18; PMID:11684447; http://dx.doi.org/10.1016/S1011-1344(01)00198-1.
14. English DR, Armstrong BK, Kricker A, Winter MG, Heenan PJ, Randell PL. Case-control study of sun exposure and squamous cell carcinoma of the skin. Int J Cancer 1998; 77:347-53; PMID:9663594; http:// dx.doi.org/10.1002/(SICI)1097-0215(19980729)77:3<347::AID-IJC7>3.0.CO;2-O.
15. Rosso S, Zanetti R, Martinez C, Tormo MJ, Schraub S, Sancho-Garnier H, et al. The multicentre south
European study 'Helios'. II: Different sun exposure patterns in the aetiology of basal cell and squamous cell carcinomas of the skin. Br J Cancer 1996; 73:1447-54; PMID:8645596; http://dx.doi.org/10.1038/ bjc.1996.275.
16. Holick MF, McLaughlin JA, Clark MB, et al. Factors that influence the cutaneous photosynthesis of previtamin D3. Science (New York, NY 1981. 211:590-593.
17. Holick MF, McLaughlin JA, Clark MB, et al. Photosynthesis of previtamin D3 in human and the physiologic consequences. Science (New York, NY 1980. 210:203-205.
18. Holick MF, Richtand NM, McNeill SC, Holick SA, Frommer JE, Henley JW, et al. Isolation and identification of previtamin D3 from the skin of rats exposed to ultraviolet irradiation. Biochemistry 1979; 18:1003-8; PMID:218609; http://dx.doi.org/10.1021/bi00573a011.
19. Webb AR, Kline L, Holick MF. Influence of season and latitude on the cutaneous synthesis of vitamin D3: exposure to winter sunlight in Boston and Edmonton will not promote vitamin D3 synthesis in human skin. J Clin Endocrinol Metab 1988; 67:373-8; PMID:2839537; http://dx.doi.org/10.1210/jcem-67-2-373.
20. Xie Z, Munson SJ, Huang N, Portale AA, Miller WL, Bikle DD. The mechanism of 1,25-dihydroxyvitamin D(3) autoregulation in keratinocytes. J Biol Chem 2002; 277:36987-90; PMID:11956203; http://dx.doi. org/10.1074/jbc.M201404200.
21. Bikle DD, Pillai S, Gee E, Hincenbergs M. Regulation of 1,25-dihydroxyvitamin D production in human keratinocytes by interferon-gamma. Endocrinology 1989; 124:655-60; PMID:2463903; http://dx.doi. org/10.1210/endo-124-2-655.
22. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell 1995; 83:835-9; PMID:8521507; http://dx.doi.org/10.1016/0092-8674(95)90199-X.
23. Haussler MR, Jurutka PW, Mizwicki M, et al. Vitamin D receptor (VDR)-mediated actions of 1alpha,25(OH) (2)vitamin D(3): genomic and non-genomic mechanisms. Best practice & research 2011. 25:543-559.
24. Battaglia S, Maguire O, Campbell MJ. Transcription factor co-repressors in cancer biology: roles and targeting. Int J Cancer 2010; 126:2511-9; PMID:20091860.
25. Pike JW, Meyer MB. The vitamin D receptor: new paradigms for the regulation of gene expression by 1,25-dihydroxyvitamin D(3). [table of contents.]. Endocrinol Metab Clin North Am 2010; 39:255-69; PMID:20511050; http://dx.doi.org/10.1016/j.ecl.2010.02.007.
26. Carlberg C, Seuter S, Heikkinen S. The first genome-wide view of vitamin D receptor locations and their mechanistic implications. Anticancer Res 2012; 32:271-82; PMID:22213316.
- '' In VitaminDWiki Vitamin D Receptor locations – Dec 2012''
27. Quigley DA, To MD, Pérez-Losada J, Pelorosso FG, Mao JH, Nagase H, et al. Genetic architecture of mouse skin inflammation and tumour susceptibility. Nature 2009; 458:505-8; PMID:19136944; http:// dx.doi.org/10.1038/nature07683.
28. Hsu JY, Feldman D, McNeal JE, Peehl DM. Reduced 1alpha-hydroxylase activity in human prostate cancer
cells correlates with decreased susceptibility to 25-hydroxyvitamin D3-induced growth inhibition. Cancer Res 2001; 61:2852-6; PMID:11306457.
29. Brozyna AA, Józwicki W, Janjetovic Z, Slominski AT. Expression of the vitamin D-activating enzyme 1a-hydroxylase (CYP27B1) decreases during melanoma progression. Hum Pathol 2012; [Epub ahead of print]. PMID:22995334; http://dx.doi.org/10.1016/j.humpath.2012.03.031.
30. Anderson MG, Nakane M, Ruan X, Kroeger PE, Wu-Wong JR. Expression of VDR and CYP24A1 mRNA in human tumors. Cancer Chemother Pharmacol 2006; 57:234-40; PMID:16180015; http://dx.doi. org/10.1007/s00280-005-0059-7.
31. Solomon C, White JH, Kremer R. Mitogen-activated protein kinase inhibits 1,25-dihydroxyvitamin D3-dependent signal transduction by phosphorylating human retinoid X receptor alpha. J Clin Invest 1999; 103:1729-35; PMID:10377179; http://dx.doi.org/10.1172/JCI6871.
32. Larriba MJ, Martín-Villar E, García JM, Pereira F, Peña C, de Herreros AG, et al. Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogenesis 2009; 30:1459-68; PMID:19502595; http://dx.doi.org/10.1093/carcin/bgp140.
33. Mittal MK, Myers JN, Misra S, Bailey CK, Chaudhuri G. In vivo binding to and functional repression of the VDR gene promoter by SLUG in human breast cells. Biochem Biophys Res Commun 2008; 372:30-4; PMID:18485278; ttp://dx.doi.org/10.1016/j.bbrc.2008.04.187.
34. Marik R, Fackler M, Gabrielson E, Zeiger MA, Sukumar S, Stearns V, et al. DNA methylation-related vitamin D receptor insensitivity in breast cancer. Cancer Biol Ther 2010; 10:44-53; PMID:20431345; http://dx.doi.org/10.4161/cbt.10.L11994.
35. Mohri T, Nakajima M, Takagi S, Komagata S, Yokoi T. MicroRNA regulates human vitamin D receptor. Int J Cancer 2009; 125:1328-33; PMID:19437538; http://dx.doi.org/10.1002/ijc.24459.
36. Essa S, Reichrath S, Mahlknecht U, Montenarh M, Vogt T, Reichrath J. Signature of VDR miRNAs and epigenetic modulation of vitamin D signaling in melanoma cell lines. Anticancer Res 2012; 32:383-9; PMID:22213330.
37. Fleet JC, DeSmet M, Johnson R, Li Y. Vitamin D and cancer: a review of molecular mechanisms. Biochem J 2012; 441:61-76; PMID:22168439; http://dx.doi.org/10.1042/BJ20110744.
38. Welsh J. Cellular and molecular effects of vitamin D on carcinogenesis. Arch Biochem Biophys 2012; 523:107-14; PMID:22085499; http://dx.doi.org/10.1016/j.abb.2011.10.019.
39. Hager G, Formanek M, Gedlicka C, Thurnher D, Knerer B, Kornfehl J. 1,25(OH)2 vitamin D3 induces elevated expression of the cell cycle-regulating genes P21 and P27 in squamous carcinoma cell lines of the head and neck. Acta Otolaryngol 2001; 121:103-9; PMID:11270487; http://dx.doi. org/10.1080/000164801300006353.
40. Swami S, Raghavachari N, Muller UR, Bao YP, Feldman D. Vitamin D growth inhibition of breast cancer cells: gene expression patterns assessed by cDNA microarray. Breast Cancer Res Treat 2003; 80:49-62; PMID:12889598; http://dx.doi.org/10.1023/A:1024487118457.
41. Lin R, Nagai Y, Sladek R, et al. Expression profiling in squamous carcinoma cells reveals pleiotropic effects of vitamin D3 analog EB1089 signaling on cell proliferation, differentiation, and immune system regulation. Molecular endocrinology (Baltimore, Md 2002. 16:1243-1256.
42. Peehl DM, Shinghal R, Nonn L, Seto E, Krishnan AV, Brooks JD, et al. Molecular activity of 1,25-dihydroxyvitamin D3 in primary cultures of human prostatic epithelial cells revealed by cDNA microarray analysis. J Steroid Biochem Mol Biol 2004; 92:131-41; PMID:15555907; http://dx.doi. org/10.1016/j.jsbmb.2004.07.003.
43. Burgering BM. A brief introduction to FOXOlogy. Oncogene 2008; 27:2258-62; PMID:18391968; http://dx.doi.org/10.1038/onc.2008.29.
44. An BS, Tavera-Mendoza LE, Dimitrov V, Wang X, Calderon MR, Wang HJ, et al. Stimulation of Sirt1-regulated FoxO protein function by the ligand-bound vitamin D receptor. Mol Cell Biol 2010; 30:4890900; PMID:20733005; http://dx.doi.org/10.1128/MCB.00180-10.
45. Morrish F, Isern N, Sadilek M, Jeffrey M, Hockenbery DM. c-Myc activates multiple metabolic networks to generate substrates for cell-cycle entry. Oncogene 2009; 28:2485-91; PMID:19448666; http://dx.doi. org/10.1038/onc.2009.112.
46. Rohan JN, Weigel NL. 1Alpha,25-dihydroxyvitamin D3 reduces c-Myc expression, inhibiting proliferation and causing G1 accumulation in C4-2 prostate cancer cells. Endocrinology 2009; 150:2046-54; PMID:19164469; http://dx.doi.org/10.1210/en.2008-1395.
47. Salehi-Tabar R, Nguyen-Yamamoto L, Tavera-Mendoza LE, Quail T, Dimitrov V, An BS, et al. Vitamin D receptor as a master regulator ofthe c-MYC/MXD1 network. Proc Natl Acad Sci U S A 2012; 109:1882732; PMID:23112173; http://dx.doi.org/10.1073/pnas.1210037109.
48. Colston KW, Perks CM, Xie SP, Holly JM. Growth inhibition of both MCF-7 and Hs578T human breast cancer cell lines by vitamin D analogues is associated with increased expression of insulin-like growth factor binding protein-3. J Mol Endocrinol 1998; 20:157-62; PMID:9513092; http://dx.doi.org/10.1677/ jme.0.0200157.
49. Sprenger CC, Peterson A, Lance R, Ware JL, Drivdahl RH, Plymate SR. Regulation of proliferation of prostate epithelial cells by 1,25-dihydroxyvitamin D3 is accompanied by an increase in insulin-like growth factor binding protein-3. J Endocrinol 2001; 170:609-18; PMID:11524241; http://dx.doi.org/10.1677/ joe.0.1700609.
50. Yang L, Yang J, Venkateswarlu S, Ko T, Brattain MG. Autocrine TGFbeta signaling mediates vitamin D3 analog-induced growth inhibition in breast cells. J Cell Physiol 2001; 188:383-93; PMID:11473365; http://dx.doi.org/10.1002/jcp.1125.
51. Zhang X, Li P, Bao J, Nicosia SV, Wang H, Enkemann SA, et al. Suppression of death receptor-mediated apoptosis by 1,25-dihydroxyvitamin D3 revealed by microarray analysis. J Biol Chem 2005; 280:3545868; PMID:16093247; http://dx.doi.org/10.1074/jbc.M506648200.
52. Lambert JR, Kelly JA, Shim M, Huffer WE, Nordeen SK, Baek SJ, et al. Prostate derived factor in human prostate cancer cells: gene induction by vitamin D via a p53-dependent mechanism and inhibition of prostate cancer cell growth. J Cell Physiol 2006; 208:566-74; PMID:16741990; http://dx.doi.org/10.1002/jcp.20692.
53. Krishnan AV, Shinghal R, Raghavachari N, Brooks JD, Peehl DM, Feldman D. Analysis of vitamin D-regulated gene expression in LNCaP human prostate cancer cells using cDNA microarrays. Prostate 2004; 59:243-51; PMID:15042599; http://dx.doi.org/10.1002/pros.20006.
54. Kodach LL, Wiercinska E, de Miranda NF, Bleuming SA, Musler AR, Peppelenbosch MP, et al. The bone morphogenetic protein pathway is inactivated in the majority of sporadic colorectal cancers. Gastroenterology 2008; 134:1332-41; PMID:18471510; http://dx.doi.org/10.1053/j.gastro.2008.02.059.
55. Lee HJ, Liu H, Goodman C, Ji Y, Maehr H, Uskokovic M, et al. Gene expression profiling changes induced by a novel Gemini Vitamin D derivative during the progression of breast cancer. Biochem Pharmacol 2006; 72:332-43; PMID:16737686; http://dx.doi.org/10.1016/j.bcp.2006.04.030.
56. Schneikert J, Behrens J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut 2007; 56:417-25; PMID:16840506; http://dx.doi.org/10.1136/gut.2006.093310.
57. Pálmer HG, González-Sancho JM, Espada J, Berciano MT, Puig I, Baulida J, et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition ofbeta-catenin signaling. J Cell Biol 2001; 154:369-87; PMID:11470825; http://dx.doi.org/10.1083/jcb.200102028.
58. Shah S, Islam MN, Dakshanamurthy S, Rizvi I, Rao M, Herrell R, et al. The molecular basis of vitamin D receptor and beta-catenin crossregulation. Mol Cell 2006; 21:799-809; PMID:16543149; http://dx.doi. org/10.1016/j.molcel.2006.01.037.
59. Aguilera O, Peña C, García JM, Larriba MJ, Ordóñez-Morán P, Navarro D, et al. The Wnt antagonist DICKKOPF-1 gene is induced by 1alpha,25-dihydroxyvitamin D3 associated to the differentiation of human colon cancer cells. Carcinogenesis 2007; 28:1877-84; PMID:17449905; http://dx.doi.org/10.1093/ carcin/bgm094.
60. Alvarez-Diaz S, Larriba MJ, Lopez-Otin C, et al. Vitamin D: Proteases, protease inhibitors and cancer. Cell cycle (Georgetown, Tex 2010. 9:32-37.
61. Ordóñez-Morán P, Larriba MJ, Pálmer HG, Valero RA, Barbáchano A, Duñach M, et al. RhoA-ROCK and p38MAPK-MSK1 medíate vitamin D effects on gene expression, phenotype, and Wnt pathway in colon cancer cells. J Cell Biol 2008; 183:697-710; PMID:19015318; http://dx.doi.org/10.1083/jcb.200803020.
62. Bikle DD, Xie Z, Tu CL. Calcium regulation ofkeratinocyte differentiation. Expert review of endocrinology & metabolism 2012. 7:461-472.
63. Peterlik M, Grant WB, Cross HS. Calcium, vitamin D and cancer. Anticancer Res 2009; 29:3687-98; PMID:19667166.
64. Díaz GD, Paraskeva C, Thomas MG, Binderup L, Hague A. Apoptosis is induced by the active metabolite
of vitamin D3 and its analogue EB1089 in colorectal adenoma and carcinoma cells: possible implications for prevention and therapy. Cancer Res 2000; 60:2304-12; PMID:10786699.
65. Blutt SE, McDonnell TJ, Polek TC, Weigel NL. Calcitriol-induced apoptosis in LNCaP cells is blockedby overexpression of Bcl-2. Endocrinology 2000; 141:10-7; PMID:10614618; http://dx.doi.org/10.1210/ en.141.1.10.
66. Pálmer HG, Sánchez-Carbayo M, Ordóñez-Morán P, Larriba MJ, Cordón-Cardó C, Muñoz A. Genetic signatures of differentiation induced by 1alpha,25-dihydroxyvitamin D3 in human colon cancer cells. Cancer Res 2003; 63:7799-806; PMID:14633706.
67. Kasiappan R, Shen Z, Tse AK, Jinwal U, Tang J, Lungchukiet P, et al. 1,25-Dihydroxyvitamin D3 Suppresses Telomerase Expression and Human Cancer Growth through MicroRNA-498. J Biol Chem 2012; 287:41297-309; PMID:23055531; http://dx.doi.org/10.1074/jbc.M112.407189.
68. Kallay E, Pietschmann P, Toyokuni S, Bajna E, Hahn P, Mazzucco K, et al. Characterization of a vitamin D receptor knockout mouse as a model of colorectal hyperproliferation and DNA damage. Carcinogenesis 2001; 22:1429-35; PMID:11532865; http://dx.doi.org/10.1093/carcin/22.9.1429.
69. Fedirko V, Bostick RM, Long Q, Flanders WD, McCullough ML, Sidelnikov E, et al. Effects ofsupplemental vitamin D and calcium on oxidative DNA damage marker in normal colorectal mucosa: a randomized clinical trial. Cancer Epidemiol Biomarkers Prev 2010; 19:280-91; PMID:20056649; http://dx.doi. org/10.1158/1055-9965.EPI-09-0448.
70. Jiang F, Li P, Fornace AJ Jr., Nicosia SV, Bai W. G2/M arrest by 1,25-dihydroxyvitamin D3 in ovarian cancer cells mediated through the induction of GADD45 via an exonic enhancer. J Biol Chem 2003; 278:48030-40; PMID:14506229; http://dx.doi.org/10.1074/jbc.M308430200.
71. Müller-Decker K, Fürstenberger G. The cyclooxygenase-2-mediated prostaglandin signaling is causally related to epithelial carcinogenesis. Mol Carcinog 2007; 46:705-10; PMID:17546626; http://dx.doi. org/10.1002/mc.20326.
72. Moreno J, Krishnan AV, Swami S, Nonn L, Peehl DM, Feldman D. Regulation of prostaglandin metabolism by calcitriol attenuates growth stimulation in prostate cancer cells. Cancer Res 2005; 65:7917-25; PMID:16140963.
73. Mantell DJ, Owens PE, Bundred NJ, Mawer EB, Canfield AE. 1 alpha,25-dihydroxyvitamin D(3) inhibits angiogenesis in vitro and in vivo. Circ Res 2000; 87:214-20; PMID:10926872; http://dx.doi.org/10.1161/01.RES.87.3.214.
74. Campbell CL, Savarese DM, Quesenberry PJ, Savarese TM. Expression of multiple angiogenic cytokines in cultured normal human prostate epithelial cells: predominance of vascular endothelial growth factor. Int J Cancer 1999; 80:868-74; PMID:10074920; http://dx.doi.org/10.1002/(SICI)1097-0215(19990315)80:6<868::AID-IJC12>3.0.CO;2-1.
75. Ben-Shoshan M, Amir S, Dang DT, Dang LH, Weisman Y, Mabjeesh NJ. 1alpha,25-dihydroxyvitamin D3 (Calcitriol) inhibits hypoxia-inducible factor-1/vascular endothelial growth factor pathway in human cancer cells. Mol Cancer Ther 2007; 6:1433-9; PMID:17431122; http://dx.doi.org/10.1158/1535-7163. MCT-06-0677.
76. Chung I, Han G, Seshadri M, Gillard BM, Yu WD, Foster BA, et al. Role of vitamin D receptor in the antiproliferative effects of calcitriol in tumor-derived endothelial cells and tumor angiogenesis in vivo. Cancer Res 2009; 69:967-75; PMID:19141646; http://dx.doi.org/10.1158/0008-5472.CAN-08-2307.
77. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science (New York, NY 2011. 331:1565-1570.
78. Zavos G, Karidis NP, Tsourouflis G, Bokos J, Diles K, Sotirchos G, et al. Nonmelanoma skin cancer after renal transplantation: a single-center experience in 1736 transplantations. Int J Dermatol 2011; 50:1496500; PMID:21790552; http://dx.doi.org/10.1111/j.1365-4632.2011.04939.x.
79. Zinser GM, Sundberg JP, Welsh J. Vitamin D(3) receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis 2002; 23:2103-9; PMID:12507934; http://dx.doi.org/10.1093/carcin/23.12.2103.
80. Indra AK, Castaneda E, Antal MC, Jiang M, Messaddeq N, Meng X, et al. Malignant transformation of DMBA/TPA-induced papillomas and nevi in the skin of mice selectively lacking retinoid-X-receptor alpha in epidemial keratinocytes. J Invest Dermatol 2007; 127:1250-60; PMID:17301838; http://dx.doi. org/10.1038/sj.jid.5700672.
81. Ellison TI, Smith MK, Gilliam AC, MacDonald PN. Inactivation of the vitamin D receptor enhances susceptibility of murine skin to UV-induced tumorigenesis. J Invest Dermatol 2008; 128:2508-17; PMID:18509362; http://dx.doi.org/10.1038/jid.2008.131.
82. Teichert AE, Elalieh H, Elias PM, Welsh J, Bikle DD. Overexpression of hedgehog signaling is associated with epidermal tumor formation in vitamin D receptor-null mice. J Invest Dermatol 2011; 131:2289-97; PMID:21814234; http://dx.doi.org/10.1038/jid.2011.196.
83. Daya-Grosjean L, Sarasin A. The role of UV induced lesions in skin carcinogenesis: an overview of oncogene and tumor suppressor gene modifications in xeroderma pigmentosum skin tumors. Mutat Res 2005; 571:43-56; PMID:15748637; http://dx.doi.org/10.1016/j.mrfmmm.2004.11.013.
84. Aszterbaum M, Rothman A, Johnson RL, Fisher M, Xie J, Bonifas JM, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol 1998; 110:885-8; PMID:9620294; http://dx.doi.org/10.1046/j.1523-
1747.1998.00222.x.
85. Pálmer HG, Anjos-Afonso F, Carmeliet G, Takeda H, Watt FM. The vitamin D receptor is a Wnt effector that controls hair follicle differentiation and specifies tumor type in adult epidermis. PLoS One 2008; 3:e1483; PMID:18213391; http://dx.doi.org/10.1371/journal.pone.0001483.
86. Schauber J, Dorschner RA, Coda AB, Büchau AS, Liu PT, Kiken D, et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest 2007; 117:803-11; PMID:17290304; http://dx.doi.org/10.1172/JCI30142.
87. Muehleisen B, Bikle DD, Aguilera C, et al. PTH/PTHrP and Vitamin D Control Antimicrobial Peptide Expression and Susceptibility to Bacterial Skin Infection. Science translational medicine 2012. 4:135ra166.
88. Pillai S, Bikle DD. Role of intracellular-free calcium in the cornified envelope formation of keratinocytes: differences in the mode of action of extracellular calcium and 1,25 dihydroxyvitamin D3. J Cell Physiol 1991; 146:94-100; PMID:1990023; http://dx.doi.org/10.1002/jcp.1041460113.
89. Bikle DD, Pillai S, Gee E. Squamous carcinoma cell lines produce 1,25 dihydroxyvitamin D, but fail to respond to its prodifferentiating effect. J Invest Dermatol 1991; 97:435-41; PMID:1875043; http://dx.doi. org/10.1111/1523-1747.ep12481267.
90. Hosomi J, Hosoi J, Abe E, Suda T, Kuroki T. Regulation of terminal differentiation of cultured mouse epidemial cells by 1 alpha,25-dihydroxyvitamin D3. Endocrinology 1983; 113:1950-7; PMID:6196178; http://dx.doi.org/10.1210/endo-113-6-1950.
91. Smith EL, Walworth NC, Holick MF. Effect of 1 alpha,25-dihydroxyvitamin D3 on the morphologic and biochemical differentiation of cultured human epidermal keratinocytes grown in serum-free conditions. J Invest Dermatol 1986; 86:709-14; PMID:2423618; http://dx.doi.org/10.1111/1523-1747.ep12276343.
92. McLane JA, Katz M, Abdelkader N. Effect of 1,25-dihydroxyvitamin D3 on human keratinocytes grown under different culture conditions. In Vitro Cell Dev Biol 1990; 26:379-87; PMID:2345124; http://dx.doi. org/10.1007/BF02623829.
93. Hawker NP, Pennypacker SD, Chang SM, Bikle DD. Regulation of human epidermal keratinocyte differentiation by the vitamin D receptor and its coactivators DRIP205, SRC2, and SRC3. J Invest Dermatol 2007; 127:874-80; PMID:17082781; http://dx.doi.org/10.1038/sj.jid.5700624.
94. Tu CL, Chang W, Xie Z, Bikle DD. Inactivation ofthe calcium sensing receptor inhibits E-cadherin-mediated cell-cell adhesion and calcium-induced differentiation in human epidermal keratinocytes. J Biol Chem 2008; 283:3519-28; PMID:18065418; http://dx.doi.org/10.1074/jbc.M708318200.
95. Tu CL, Oda Y, Komuves L, Bikle DD. The role ofthe calcium-sensing receptor in epidemial differentiation. Cell Calcium 2004; 35:265-73; PMID:15200150; http://dx.doi.org/10.1016/j.ceca.2003.10.019.
96. Xie Z, Bikle DD. Cloning of the human phospholipase C-gamma1 promoter and identification of a DR6-type vitamin D-responsive element. J Biol Chem 1997; 272:6573-7; PMID:9045685; http://dx.doi. org/10.1074/jbc.272.10.6573.
97. Xie Z, Bikle DD. Phospholipase C-gamma1 is required for calcium-induced keratinocyte differentiation. J Biol Chem 1999; 274:20421-4; PMID:10400667; http://dx.doi.org/10.1074/jbc.274.29.20421.
98. Xie Z, Bikle DD. Inhibition of 1,25-dihydroxyvitamin-D-induced keratinocyte differentiation by blocking the expression of phospholipase C-gamma1. J Invest Dermatol 2001; 117:1250-4; PMID:11710940; http://dx.doi.org/10.1046/j.0022-202x.2001.01526.x.
99. Matsumoto K, Hashimoto K, Nishida Y, Hashiro M, Yoshikawa K. Growth-inhibitory effects of 1,25-dihydroxyvitamin D3 on normal human keratinocytes cultured in serum-free medium. Biochem Biophys Res Commun 1990; 166:916-23; PMID:2405860; http://dx.doi.org/10.1016/0006-291X(90)90898-W.
100. Bikle DD. The vitamin D receptor: a tumor suppressor in skin. Discov Med 2011; 11:7-17; PMID:21276406.
101. Oda Y, Uchida Y, Moradian S, Crumrine D, Elias PM, Bikle DD. Vitamin D receptor and coactivators SRC2 and 3 regulate epidermis-specific sphingolipid production and permeability barrier formation. J Invest Dermatol 2009; 129:1367-78; PMID:19052561; http://dx.doi.org/10.1038/jid.2008.380.
102. Schauber J, Dorschner RA, Yamasaki K, Brouha B, Gallo RL. Control ofthe innate epithelial antimicrobial response is cell-type specific and dependent on relevant microenvironmental stimuli. Immunology 2006; 118:509-19; PMID:16895558.
103. Xie Z, Komuves L, Yu QC, Elalieh H, Ng DC, Leary C, et al. Lack of the vitamin D receptor is associated with reduced epidermal differentiation and hair follicle growth. J Invest Dermatol 2002; 118:11-6; PMID:11851870; http://dx.doi.org/10.1046/j.1523-1747.2002.01644.x.
104. Bikle DD, Elalieh H, Chang S, Xie Z, Sundberg JP. Development and progression of alopecia in the vitamin D receptor null mouse. J Cell Physiol 2006; 207:340-53; PMID:16419036; http://dx.doi.org/10.1002/jcp.20578.
105. Bikle DD, Chang S, Crumrine D, Elalieh H, Man MQ, Choi EH, et al. 25 Hydroxyvitamin D 1 alpha-hydroxylase is required for optimal epidermal differentiation and permeability barrier homeostasis. J Invest Dermatol 2004; 122:984-92; PMID:15102089; http://dx.doi.org/10.1111/j.0022-202X.2004.22424.x.
106. Barnfield PC, Zhang X, Thanabalasingham V, Yoshida M, Hui CC. Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple mechanisms. Differentiation 2005; 73:397405; PMID:16316410; http://dx.doi.org/10.1111/j.1432-0436.2005.00042.x.
107. Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergstróm A, et al. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell 2006; 10:187-97; PMID:16459298; http://dx.doi.org/10.1016/j.devcel.2005.12.013.
108. Regl G, Kasper M, Schnidar H, Eichberger T, Neill GW, Ikram MS, et al. The zinc-finger transcription factor GLI2 antagonizes contact inhibition and differentiation of human epidemial cells. Oncogene 2004; 23:1263-74; PMID:14691458; http://dx.doi.org/10.1038/sj.onc.1207240.
109. Regl G, Kasper M, Schnidar H, Eichberger T, Neill GW, Philpott MP, et al. Activation of the BCL2 promoter in response to Hedgehog/GLI signal transduction is predominantly mediated by GLI2. Cancer Res 2004; 64:7724-31; PMID:15520176; http://dx.doi.org/10.1158/0008-5472.CAN-04-1085.
110. Regl G, Neill GW, Eichberger T, Kasper M, Ikram MS, Koller J, et al. Human GLI2 and GLI1 are part of a positive feedback mechanism in Basal Cell Carcinoma. Oncogene 2002; 21:5529-39; PMID:12165851; http://dx.doi.org/10.1038/sj.onc.1205748.
111. Grachtchouk M, Mo R, Yu S, Zhang X, Sasaki H, Hui CC, et al. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat Genet 2000; 24:216-7; PMID:10700170; http://dx.doi.org/10.1038/73417.
112. Nilsson M, Undén AB, Krause D, Malmqwist U, Raza K, Zaphiropoulos PG, et al. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc Natl Acad Sci U S A 2000; 97:3438-43; PMID:10725363; http://dx.doi.org/10.1073/pnas.97.7.3438.
113. Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85:841-51; PMID:8681379; http://dx.doi.org/10.1016/S0092-8674(00)81268-4.
114. Ping XL, Ratner D, Zhang H, Wu XL, Zhang MJ, Chen FF, et al. PTCH mutations in squamous cell carcinoma of the skin. J Invest Dermatol 2001; 116:614-6; PMID:11286632; http://dx.doi.org/10.1046/j.1523-1747.2001.01301.x.
115. Uhmann A, Niemann H, Lammering B, Henkel C, Hess I, Nitzki F, et al. Antitumoral effects of calcitriol in basal cell carcinomas involve inhibition of hedgehog signaling and induction of vitamin D receptor signaling and differentiation. Mol Cancer Ther 2011; 10:2179-88; PMID:21878656; http://dx.doi. org/10.1158/1535-7163.MCT-11-0422.
116. Luderer HF, Gori F, Demay MB. Lymphoid enhancer-binding factor-1 (LEF1) interacts with the DNA-binding domain of the vitamin D receptor. J Biol Chem 2011; 286:18444-51; PMID:21471213; http:// dx.doi.org/10.1074/jbc.M110.188219.
117. Bijlsma MF, Spek CA, Zivkovic D, van de Water S, Rezaee F, Peppelenbosch MP. Repression of smoothened by patched-dependent (pro-)vitamin D3 secretion. PLoS Biol 2006; 4:e232; PMID: 16895439; http://dx.doi.org/10.1371/journal.pbio.0040232.
118. Tang JY, Xiao TZ, Oda Y, Chang KS, Shpall E, Wu A, et al. Vitamin D3 inhibits hedgehog signaling and proliferation in murine Basal cell carcinomas. Cancer Prev Res (Phila) 2011; 4:744-51; PMID:21436386; http://dx.doi.org/10.1158/1940-6207.CAPR-10-0285.
119. He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science (New York, NY 1998. 281:1509-1512.
120. Xie Z, Bikle DD. The recruitment of phosphatidylinositol 3-kinase to the E-cadherin-catenin complex at the plasma membrane is required for calcium-induced phospholipase C-gamma1 activation and human keratinocyte differentiation. J Biol Chem 2007; 282:8695-703; PMID:17242406; http://dx.doi. org/10.1074/jbc.M609135200.
121. Bienz M. beta-Catenin: a pivot between cell adhesion and Wnt signalling. Curr Biol 2005; 15:R64-7; PMID:15668160; http://dx.doi.org/10.1016/j.cub.2004.12.058.
122. Shah S, Hecht A, Pestell R, Byers SW. Trans-repression of beta-catenin activity by nuclear receptors. J Biol Chem 2003; 278:48137-45; PMID:12972427; http://dx.doi.org/10.1074/jbc.M307154200.
123. Chan EF, Gat U, McNiff JM, Fuchs E. A common human skin tumour is caused by activating mutations in beta-catenin. Nat Genet 1999; 21:410-3; PMID:10192393; http://dx.doi.org/10.1038/7747.
124. Gat U, DasGupta R, Degenstein L, Fuchs E. De Novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell 1998; 95:605-14; PMID:9845363; http://dx.doi. org/10.1016/S0092-8674(00)81631-1.
125. Xia J, Urabe K, Moroi Y, Koga T, Duan H, Li Y, et al. beta-Catenin mutation and its nuclear localization are confirmed to be frequent causes of Wnt signaling pathway activation in pilomatricomas. J Dermatol Sci 2006; 41:67-75; PMID:16378715; http://dx.doi.org/10.1016/j.jdermsci.2005.09.005.
126. Cianferotti L, Cox M, Skorija K, Demay MB. Vitamin D receptor is essential for normal keratinocyte stem cell function. Proc Natl Acad Sci U S A 2007; 104:9428-33; PMID:17517646; http://dx.doi.org/10.1073/ pnas.0702884104.
127. Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, et al. DCs metabolize sunlight-induced vitamin D3 to 'program' T cell attraction to the epidermal chemokine CCL27. Nat Immunol 2007; 8:28593; PMID:17259988; http://dx.doi.org/10.1038/ni1433.
128. Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE. Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J Immunol 2007; 179:1634-47; PMID:17641030.
129. Bikle DD. Vitamin D and immune function: understanding common pathways. Curr Osteoporos Rep 2009; 7:58-63; PMID:19631030; http://dx.doi.org/10.1007/s11914-009-0011-6.
130. Rigby WF, Stacy T, Fanger MW. Inhibition of T lymphocyte mitogenesis by 1,25-dihydroxyvitamin D3 (calcitriol). J Clin Invest 1984; 74:1451-5; PMID:6332829; http://dx.doi.org/10.1172/JCI111557.
131. Lemire JM, Archer DC, Beck L, Spiegelberg HL. Immunosuppressive actions of 1,25-dihydroxyvitamin D3: preferential inhibition of Th1 functions. J Nutr 1995; 125(Suppl):1704S-8S; PMID:7782931.
132. Daniel C, Sartory NA, Zahn N, Radeke HH, Stein JM. Immune modulatory treatment of trinitrobenzene sulfonic acid colitis with calcitriol is associated with a change of a T helper (Th) 1/Th17 to a Th2 and regulatory T cell profile. J Pharmacol Exp Ther 2008; 324:23-33; PMID:17911375; http://dx.doi.
org/10.1124/jpet.107.127209.
133. Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature 2008; 453:1051-7; PMID:18563156; http://dx.doi.org/10.1038/nature07036.
134. Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HF, O'Garra A. 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J Immunol 2001; 167:4974-80; PMID:11673504.
135. Penna G, Adorini L. 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol 2000; 164:240511; PMID:10679076.
136. Griffin MD, Xing N, Kumar R. Vitamin D and its analogs as regulators of immune activation and antigen presentation. Annu Rev Nutr 2003; 23:117-45; PMID:12651965; http://dx.doi.org/10.1146/annurev.nutr.23.011702.073114.
137. Liu PT, Krutzik SR, Modlin RL. Therapeutic implications of the TLR and VDR partnership. Trends Mol Med 2007; 13:117-24; PMID:17276732; http://dx.doi.org/10.1016/j.molmed.2007.01.006.
138. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature 2007; 449:819-26; PMID:17943118; http://dx.doi.org/10.1038/nature06246.
139. Gombart AF, Borregaard N, Koeffler HP. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J 2005; 19:1067-77; PMID:15985530; http://dx.doi.org/10.1096/fj.04-3284com.
140. Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, et al. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer ofantimicrobial peptide gene expression. J Immunol 2004; 173:2909-12; PMID:15322146.
141. Liu PT, Stenger S, Li H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science (New York, NY 2006. 311:1770-1773.
142. Kripke ML, Munn CG, Jeevan A, Tang JM, Bucana C. Evidence that cutaneous antigen-presenting cells migrate to regional lymph nodes during contact sensitization. J Immunol 1990; 145:2833-8; PMID:2212665.
143. Matsumura Y, Ananthaswamy HN. Toxic effects ofultraviolet radiation on the skin. Toxicol Appl Pharmacol 2004; 195:298-308; PMID:15020192; http://dx.doi.org/10.1016/j.taap.2003.08.019.
144. Toews GB, Bergstresser PR, Streilein JW. Epidermal Langerhans cell density determines whether contact hypersensitivity or unresponsiveness follows skin painting with DNFB. J Immunol 1980; 124:445-53; PMID:6153101.
145. Simon JC, Cruz PD Jr., Tigelaar RE, Sontheimer RD, Bergstresser PR. Adhesion molecules CD11a, CD18, and ICAM-1 on human epidermal Langerhans cells serve a functional role in the activation of alloreactive T cells. J Invest Dermatol 1991; 96:148-51; PMID:1670950; http://dx.doi.org/10.1111/1523-1747.ep12515946.
146. Tang A, Udey MC. Inhibition of epidermal Langerhans cell function by low dose ultraviolet B radiation. Ultraviolet B radiation selectively modulates ICAM-1 (CD54) expression by murine Langerhans cells. J Immunol 1991; 146:3347-55; PMID:1673983.
147. Glaser R, Navid F, Schuller W, Jantschitsch C, Harder J, Schróder JM, et al. UV-B radiation induces the expression of antimicrobial peptides in human keratinocytes in vitro and in vivo. J Allergy Clin Immunol 2009; 123:1117-23; PMID:19342087; http://dx.doi.org/10.1016/j.jaci.2009.01.043.
148. Mallbris L, Edstróm DW, Sundblad L, Granath F, Stahle M. UVB upregulates the antimicrobial protein hCAP18 mRNA in human skin. J Invest Dermatol 2005; 125:1072-4; PMID:16297211.
149. Kripke ML, Fisher MS. Immunologic parameters of ultraviolet carcinogenesis. J Natl Cancer Inst 1976; 57:211-5; PMID:1003502.
150. Dixon KM, Deo SS, Norman AW, Bishop JE, Halliday GM, Reeve VE, et al. In vivo relevance for photoprotection by the vitamin D rapid response pathway. J Steroid Biochem Mol Biol 2007; 103:451-6; PMID:17223553; http://dx.doi.org/10.1016/j.jsbmb.2006.11.016.
151. Damian DL, Kim YJ, Dixon KM, Halliday GM, Javeri A, Mason RS. Topical calcitriol protects from UV-induced genetic damage but suppresses cutaneous immunity in humans. Exp Dermatol 2010; 19:e23-30; PMID:19758324; http://dx.doi.org/10.1111/j.1600-0625.2009.00955.x.
152. Li G, Mitchell DL, Ho VC, Reed JC, Tron VA. Decreased DNA repair but normal apoptosis in ultraviolet-irradiated skin of p53-transgenic mice. Am J Pathol 1996; 148:1113-23; PMID:8644854.
153. Tlsty TD, Margolin BH, Lum K. Differences in the rates of gene amplification in nontumorigenic and tumorigenic cell lines as measured by Luria-Delbrück fluctuation analysis. Proc Natl Acad Sci U S A 1989; 86:9441-5; PMID:2687881; http://dx.doi.org/10.1073/pnas.86.23.9441.
154. Tlsty TD. Normal diploid human and rodent cells lack a detectable frequency of gene amplification. Proc Natl Acad Sci U S A 1990; 87:3132-6; PMID:2326271; http://dx.doi.org/10.1073/pnas.87.8.3132.
155. Maher VM, Dorney DJ, Mendrala AL, Konze-Thomas B, McCormick JJ. DNA excision-repair processes in human cells can eliminate the cytotoxic and mutagenic consequences of ultraviolet irradiation. Mutat Res 1979; 62:311-23; PMID:503098; http://dx.doi.org/10.1016/0027-5107(79)90087-3.
156. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006; 444:633-7; PMID:17136093; http://dx.doi.org/10.1038/nature05268.
157. Bielas JH, Loeb KR, Rubin BP, True LD, Loeb LA. Human cancers express a mutator phenotype. Proc Natl Acad Sci U S A 2006; 103:18238-42; PMID:17108085; http://dx.doi.org/10.1073/pnas.0607057103.
158. Chen RH, Maher VM, McCormick JJ. Effect of excision repair by diploid human fibroblasts on the kinds and locations of mutations induced by (+/-)-7 beta,8 alpha-dihydroxy-9 alpha,10 alpha-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene in the coding region of the HPRT gene. Proc Natl Acad Sci U S A 1990; 87:8680-4; PMID:2122466; http://dx.doi.org/10.1073/pnas.87.21.8680.
159. Wood RD. DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie 1999; 81:39-44; PMID:10214908; http://dx.doi.org/10.1016/S0300-9084(99)80036-4.
160. Sertic S, Pizzi S, Lazzaro F, et al. NER and DDR: classical music with new instruments. Cell cycle (Georgetown, Tex 2012. 11:668-674.
161. Mellon I, Bohr VA, Smith CA, Hanawalt PC. Preferential DNA repair of an active gene in human cells. Proc Natl Acad Sci U S A 1986; 83:8878-82; PMID:3466163; http://dx.doi.org/10.1073/pnas.83.23.8878.
162. Mellon I, Spivak G, Hanawalt PC. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell 1987; 51:241-9; PMID:3664636; http://dx.doi. org/10.1016/0092-8674(87)90151-6.
163. Mellon I, Rajpal DK, Koi M, et al. Transcription-coupled repair deficiency and mutations in human mismatch repair genes. Science (New York, NY 1996. 272:557-560.
164. Bohr VA. Gene specific DNA repair. Carcinogenesis 1991; 12:1983-92; PMID:1934282; http://dx.doi. org/10.1093/carcin/12.11.1983.
165. Hanawalt PC. Transcription-coupled repair and human disease. Science. New York, NY: 1994;266:1957-1958.
166. Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411:366-74; PMID:11357144; http://dx.doi.org/10.1038/35077232.
167. Wood RD, Mitchell M, Sgouros J, et al. Human DNA repair genes. Science (New York, NY 2001. 291:1284-1289.
168. Demetriou SK, Ona-Vu K, Teichert AE, Cleaver JE, Bikle DD, Oh DH. Vitamin D receptor mediates DNA repair and is UV inducible in intact epidermis but not in cultured keratinocytes. J Invest Dermatol 2012; 132:2097-100; PMID:22495177; http://dx.doi.org/10.1038/jid.2012.107.
169. Dixon KM, Deo SS, Wong G, Slater M, Norman AW, Bishop JE, et al. Skin cancer prevention: a possible role of 1,25dihydroxyvitamin D3 and its analogs. J Steroid Biochem Mol Biol 2005; 97:137-43; PMID:16039116; http://dx.doi.org/10.1016/j.jsbmb.2005.06.006.
170. Gupta R, Dixon KM, Deo SS, Holliday CJ, Slater M, Halliday GM, et al. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J Invest Dermatol 2007; 127:707-15; PMID:17170736; http://dx.doi.org/10.1038/sj.jid.5700597.
171. Moll PR, Sander V, Frischauf AM, Richter K. Expression profiling of vitamin D treated primary human keratinocytes. J Cell Biochem 2007; 100:574-92; PMID:16960875; http://dx.doi.org/10.1002/jcb.21061.