Telomeres (which extend life) appear to be extended by Vitamin D
Update: Telemeres in boys were 2.5% longer if 9 ng higher vitamin D – July 2018
Vitamin D as predictor of telomere length in the transition from child to adolescent
Revue d'Épidémiologie et de Santé Publique, Volume 66, Supplement 5, July 2018, Page S237, https://doi.org/10.1016/j.respe.2018.05.015
Van AartaN. MichelsaI.SioenabD .MartenscT.NawrotcS.De Henauwa
Results
Telomere length was shorter with increasing age (P = 0.01). Vitamin D was not associated with telomere length in cross-sectional analysis combining both sexes, but in boys higher vitamin D was related to longer telomeres. An interquartile increase in vitamin D (9.4 ng/mL) was associated with a 2.5% (95% confidence interval: 0.1 to 4.9%; P = 0.04) increase in telomere length. Longitudinal analysis showed similar results for boys (P = 0.02).
The role of telomeres and vitamin D in cellular aging and age-related diseases - March 2015
Clin Chem Lab Med. 2015 Mar 21. pii: /j/cclm.ahead-of-print/cclm-2014-1184/cclm-2014-1184.xml. doi: 10.1515/cclm-2014-1184
Irene Pusceddu, Christopher-John L. Farrell, Angela Maria Di Pierro, Erika Jani, Wolfgang Herrmann and Markus Herrmann*
Corresponding author: Associate Professor, Dr. med. habit. Markus Herrmann, Department of Clinical Pathology, District Hospital of Bolzano, 39100 Bolzano, Italy, Phone: +39 0 471 90 9675, E-mail: markusherr@aol.com; Department of Clinical Chemistry and Laboratory Medicine, Saarland University Hospital, Homburg/Saar, Germany; and Laverty Pathology, Sydney, NSW, Australia Irene Pusceddu and Wolfgang Herrmann: Department of Clinical Chemistry and Laboratory Medicine, Saarland University Hospital, Homburg/Saar, Germany
Christopher-John L. Farrell: Laverty Pathology, Sydney, NSW, Australia
Angela Maria Di Pierro and Erika Jani: Department of Clinical Pathology, District Hospital of Bolzano, Bolzano, Italy
Abstract: Aging is a complex biological process characterized by a progressive decline of organ functions leading to an increased risk of age-associated diseases and death. Decades of intensive research have identified a range of molecular and biochemical pathways contributing to aging. However, many aspects regarding the regulation and interplay of these pathways are insufficiently understood. Telomere dysfunction and genomic instability appear to be of critical importance for aging at a cellular level. For example, age-related diseases and premature aging syndromes are frequently associated with telomere shortening. Telomeres are repetitive nucleotide sequences that together with the associated sheltrin complex protect the ends of chromosomes and maintain genomic stability. Recent studies suggest that micronutrients, such as vitamin D, folate and vitamin B12, are involved in telomere biology and cellular aging. In particular, vitamin D is important for a range of vital cellular processes including cellular differentiation, proliferation and apoptosis. As a result of the multiple functions of vitamin D it has been speculated that vitamin D might play a role in telomere biology and genomic stability. Here we review existing knowledge about the link between telomere biology and cellular aging with a focus on the role of vitamin D. We searched the literature up to November 2014 for human studies, animal models and in vitro experiments that addressed this topic.
📄 Download the PDF will all of the images from VitaminDWiki
Introduction
Aging is a multifactorial biological process characterized by a progressive decline of organ functions leading to an increased risk of age-associated diseases and death. Deterioration of genomic integrity and genomic instability are critical aspects in aging at a cellular level. Telomeres appear to be of critical importance for genomic stability and cellular aging. Telomeres are the end caps of chromosomes and were first identified in 1938 by Hermann Muller [1, 2]. Since then telomere biology has been widely investigated and numerous studies indicate an involvement of telomeres in the process of aging [3]. Telomere shortening and dysfunction have been proposed as indicators of cellular aging and are associated with age-related diseases including cardiovascular disease (CVD), type 2 diabetes mellitus (T2DM), cancer or chronic obstructive pulmonary disease [3]. Micronutrients, such as vitamins and trace elements play an important role in cell metabolism and some studies suggest a direct effect of these micronutrients on telomere biology and cellular aging [4]. Vitamin D, for example, is a steroid hormone with genomic and non-genomic activities that is involved in the regulation of cell proliferation, differentiation and apoptosis [5]. As a result of the multiple functions of vitamin D it has been speculated that vitamin D might play a role in telomere biology and genomic stability. Here we review existing knowledge about the potential link between vitamin D and telomere biology with a focus on age-related diseases.
Cellular aging
Aging and senescence
Aging is a physiological condition characterized by a progressive decline of organ function ultimately leading to death [6]. Several molecular and biochemical pathways contribute to aging and one of the most important of these is cellular senescence [7]. Cellular senescence is an irreversible arrest of cell proliferation that can be induced in different ways including genomic damage, toxins, irradiation, oxidative stress, oncogene expression, tumor suppressor gene activation and epigenomic alterations [8]. The state of senescence is established and maintained by at least two major tumor suppressor pathways: the p53/ p21 and the p16INK4a/pRB pathways [8]. The p53/p21 pathway is activated by genomic or epigenomic stressors through the activation of the DNA damage response (DDR) [8]. The DDR is a network of cellular pathways that sense, signal and repair DNA lesions [9]. It prevents the generation of potentially deleterious mutations and avoids genomic instability and dysfunction [9]. Stress that does not entail direct genomic damage can induce p16INK4a expression, which activates the pRB tumor suppressor, that in turn silences certain pro-proliferative genes [7, 8]. Activation of both, p53/p21 and p16INK4a/pRB, triggers a signaling cascade that induces apoptosis and/or senescence [8]. The nature and degree of stress as well as the cell type, the balance between pro-senescent and pro- apoptotic pathways also decide cell fate [10].
A range of biochemical features characterizes senescent cells: they are metabolically active, relatively resistant to apoptosis and also secrete pro-inflammatory cytokines, chemokines and proteases leading to a chronic inflammatory condition [7, 8]. This phenotype is known as senescence-associated secretory phenotype (SASP) [8]. Proteins that are associated with SASP are tumor necrosis factor a (TNF-a), interleukin 6 (IL-6), matrix metallo- proteinases (MMPs), monocyte chemoattractant protein-1 (MCP-1) and insulin-like growth factor binding proteins (IGFBPs) [8]. In addition, an intracellular IL-1a/miR- 146a/b/IL-6/CCAAT/enhancer binding protein (C/EBP-b) loop as well as related p38/nuclear factor K-light chain enhancer of activated B cells (NF-kB) - and mammalian target for rapamycin (mTOR) - mediated pathways appear to contribute to the SASP phenotype [8] . Moreover IL-6 and IL-8 are able to stimulate or inhibit Wnt (wingless, Drosophila segment polarity gene and abd integrated, vertebrate homolog) signaling and cell proliferation, respectively, depending on the physiological context .8]. The Wnt signaling pathway regulates crucial aspects of cell fate determination, cell migration, cell polarity, neural patterning and organogenesis during embryonic development [11]. Cellular aging is also influenced by endocrine factors, like insulin-like growth factor 1 (IGF-1), Klotho and fibroblast growth factor 23 (FGF-23) . 12]. Reduced IGF-1 expression in mice dramatically prolongs the lifespan, probably due to the regulation of forkhead box transcription factor 1 (FOXO1) activity [12]. The Klotho-FGF23 axis is a well known aging network; in fact, overexpression of Klotho in mice extends lifespan [13-15].
Telomere biology
Telomere dysfunction is one of the most important oncogenic stimuli able to activate a senescence response [8]. Telomeres (from Greek telos ‘end’ and meros ‘part’) are nucleoprotein structures that are highly conserved and are the end caps of chromosomes [2]. In humans, the telo- meric DNA sequence is a tandem repeat of six nucleotides -TTAGGG - that extends approximately 10 -15 kilobases [16, 17]. Telomeric DNA is double-stranded for most of its length with a 3’G rich single-stranded overhang of several repeats at the very end (150-200 bp) [16-20] (Figure 1A). The G-strand overhang can also fold back and anneal with the double-stranded region of telomeres forming a looped structure known as a T-loop [18-20] (Figure 1B). The closed configuration of the T-loop provides a protective cap that defines the natural end of the chromosome
. Telomeres participate in the maintenance of genomic and cellular stability and replication; in fact, they protect the genome from degradation, unwanted recombination and chromosomal fusion [18-20] . Chromosomal fusion can lead to chromosome imbalance, gene amplification, non-reciprocal translocations and altered gene expression
. In addition, the folding of the telomeric sequence in a three-dimensional (3D) structure protects telomeres from the recognition of the 3 . OH as a double strand break-like (DSB) . 19] . In the presence of telomere dysfunction all these conditions would activate the DDR cascade leading to cellular senescence [21].
Telomere repeats are bound to telomere-specific proteins, as shown in Figure 1A, B and C. Some of these proteins are associated exclusively with telomeres and compose the telomeric core complex known as telosome or sheltrin [22, 23]. The telomere sheltrin is composed of six telomere-specific proteins [22, 23]: Telomeric Repeat binding Factor 1 (TRF1), Telomeric Repeat binding Factor 2 (TRF2), Repressor/Activator Protein 1 (RAP1),
Figure 1: Telomere structure.
(A) Telomers are composed by a double strand region of -TTAGGG- repetitions and by a single strand region called G-strand overhang. Two protein complexes are bound to telomeres, the telomere repeat binding factor 1 (TRF1) complex and the telomere repeat binding factor 2 (TRF2) complex. (B) The G-strand overhand can fold back and invades the double strand region leading to the formation of T-loop and D-loop structures. The resulting 3D conformation protects the 3'OH end of the chromosome. (C) Composition of the two main telomere-associated protein complexes. The TRF1 complex is involved in telomere length control, whereas the TRF2 complex functions as protective end cap of telomeres. Modified from Blasco et al. [18, 19].
TRF1 Interacting protein 1 (TIN2), TINT1/PIP1/PTOP 1 (TPP1) and Protection Of Telomeres 1 (POT1). The TRF1 complex is involved in telomere length control regulating the access of the telomerase to the telomere sequence [24]. The TRF2 complex is thought to have a fundamental role in protecting the G-strand overhang from degradation, as well as in preventing telomeric fusions [22] (Figure 1C).
In vitro culture of somatic cells provides evidence of a limited replicative potential. This phenomenon was originally described by Hayflick in human fibroblasts, serially passaged in culture [25]. The Hayflick limit of cellular division is the maximum number of population doublings cells may undergo and is mediated by telomere erosion. Due to the inability of the DNA polymerase to maintain the length of the 3 ' overhang, somatic cell replication is accompanied by a loss of 50-200 bp of telomeric sequence at every cell division [26]. The consequence of this phenomenon is that a somatic cell can undergo a defined number of doublings before telomeres become critically short, lose their protective properties and send cells into senescence, or cause cell death [23]. In contrast to most somatic cells, hematopoietic stem cells, keratinocytes in the basal layer of the epidermis, uterine endometrial cells, germ cells and various tumors avoid telomere shortening by activation of telomerase [26, 27]. Telomerase is a ribo- nucleoprotein with reverse transcription activity, which adds de novo telomere hexanucleotide repeats to the chromosome ends [18, 19, 28, 29] (Figure 2A and B ). Telomerase contains a highly conserved reverse transcriptase [human telomerase reverse transcriptase (hTERT)], an associated template RNA [telomerase RNA component (TERC )] and a key auxiliary protein known as Dyskerin [28, 29]. Exogenous expression of telomerase in primary human fibroblasts is sufficient to reconstitute telomerase activity and to counteract telomere erosion. The resulting telomere maintenance immortalizes most human cell types [24].
Telomere length - analytical aspects
To date a range of different methods for the measurement of telomere length exists. Each method has advantages and disadvantages. The first method that has been developed is based on the Southern blot technique [30]. It relies on the absence of restriction enzyme recognition sites within the TTAGGG tandem repeat sequence. Genomic DNA is digested with cutting restriction endonucleases, like Hinfl and Rsal to degrade the non-telom- eric sequence. The terminal restriction fragments (TRFs) are separated by gel electrophoresis and transferred to a membrane. A radioactively-labeled telomere-specific
Figure 2: Telomerase structure and activity.
(A) The telomerase enzyme is composed by the human telomerase reverse transcriptase (hTERT), the telomerase RNA component (TERC) and the key auxiliary protein like Dyskerin, NOP10 (novel nucleolar protein 10), GAR1 (glycine and arginine rich domain) and NHP2 (non-histone protein 2). (B) Telomerase adds de novo telomere hexanucleotide repeats to the ends of the chromosome in a three-stage process: 1) recognition and binding of the hTERT complex; 2) elongation by adding complementary nucleotides;
3) translocation of the hTERT complex. Stages 2 and 3 are then repeated. Modified from Smogorzewska et al. and Marrone et al. [24,29].
probe is then used for hybridization and telomere length is evaluated by densitometry. The result of this analysis indicates the distribution of telomere length over all chomosomes and cells present in the sample [30]. It does not provide information about telomere length in individual cells or individual chromosomes. The method requires a substantial quantity of DNA (i.e., pg) is time consuming and not suitable for large population-based studies. The analytical principle of this hybridization- based method implies a decreasing signal as telomerebecome shorter. There is a threshold telomere length below which no detectable signal will be generated. The inability to detect very short telomeres limits the utility of this method in studies of cellular aging, where short telomeres are of particular interest [30]. Although the TRFs assay is considered the gold standard, the above mentioned disadvantages have led to the development of other methods that overcome the above mentioned limitations. Quantitative fluorescence in situ hybridization (Q-FISH) is an alternative method that was initially described by Lansdorp et al. [30]. In this method dividing cells are arrested in metaphase and are then fixed, permeabilized and stained with a fluorescent probe that binds specifically to the telomeric sequence. The intensity of the fluorescent signal at each telomere is a measure of telomere length. The main advantage of this method is the capability to measure telomere length of individual cells. The combination of Q-FISH with conventional FISH using chromosome-specific probes, allows not only the analysis of telomere length of individual cells, but also of individual chromosomes [30]. Similarly to the Southern blot-based method described above, Q-FISH is also a hybridization technique, which implies the existence of a threshold telomere length below which no detectable signal is produced. As Q-FISH is performed on cells in metaphase, the analysis is limited to cells that proliferate in vitro. Consequently, not all cells are suitable to be analyzed with this method. Recently, a confocal telomere quantitative fluorescence in situ hybridization method (Telomapping) has been described [31]. This method allows the quantification of telomere length in tissue samples or biopsies and gradients of telomere length within the tissue can be studied. The longest telomeres have been found in the stem cell compartment. Canela et al. described another Q-FISH-based method [32]. This automated high-throughput Q-FISH (HT Q-FISH) method combines the Q-FISH technology of telomeres in interphase nuclei with an automated high-throughput (HT) microscopy. It allows the quantification of telomere length as well as the percentage of short telomeres in large sample sets [32]. Another powerful variant of Q-FISH is the flow-FISH, where interphase telomere FISH is coupled with flow-cytometry [30]. Flow-FISH measures the total telomeric signal of a cell, which is the sum of all telomeres in each cell. As it is a flow-cytometry- based technique, telomere length on a large number of individual cells can be performed allowing the analysis of telomere length of defined populations or subpopulations of cells. As this method requires vital cells, it is widely used for the analysis of telomere length in hematopoietic cells. Finally, a quantitative-polymerase chain reaction (Q-PCR) based assay for telomere length analysis has been described [33]. This method requires the amplification of two DNA regions by PCR. The first region is the telomeric sequence (telo) that is recognized by specific primers. The second region is a single copy gene (SCG) that is used as control for amplification. The most frequent SCG used is the 36B4 gene, which encodes the acidic ribosomal phosphoprotein P0. After amplification, telomere length is quantified as the amount of telomeric sequence compared to SCG and is expressed as a relative telomere length or T/S ratio [33]. This method allows a relative quantification of the telomeric sequence present in all cells of the sample. As for TRFs assay, it is not possible to evaluate telomere length of individual cells or chromosomes. However, the Q-PCR-based technique is fast, highly sensitive and requires low amounts of DNA (i.e., nanograms). As a result of these characteristics it is often chosen for large population-based studies. Further adaptations of this method are the multiplex- based assay, in which both DNA regions are amplified in the same reaction, reducing also the amount of DNA [34]; or the absolute telomere length analysis, in which a synthetized oligomer standard for both DNA regions is co-amplified allowing the expression of telomere length as an absolute value [35].
The intra- and inter- laboratory variability, the lack of a common standard reference and the absence of reference ranges, are limiting the application of telomere length analysis in clinical laboratories. Recently, an international collaborative study group has been developed in order to harmonize telomere length analysis between laboratories [36] .
Telomeres and age-associated diseases
With increasing age most human somatic tissues and adult stem cells undergo telomere attrition, as they do not express sufficient amounts of telomerase to maintain telomere length indefinitely [37]. Dysfunctional telomeres may also arise by an independent mechanism called telomere uncapping [38] . In this alternative process, there is interference between the telomeric sequence and telomere-binding proteins, frequently the result of mutations, which leads to immediate uncapping of telomeres without telomere shortening [38]. Both critically short telomeres and uncapped telomeres impair cell viability and lead to senescence or apoptosis [37, 38].
A number of age-related conditions, like CVD, T2DM, neurodegenerative diseases and premature aging syndromes (e.g., congenital dyskeratosis), are characterized by a faster-than-normal rate of telomere shortening [3, 7, 39-41]. However, the association between telomere length and age-associated diseases is still a matter of debate. Some prospective studies have shown that short telomeres are associated with increased all-cause mortality [42-46], whereas other studies have not found such an association [47-51].
CVD is among the most frequent age-related disease and the number one cause of death. There is substantial evidence linking CVD with telomere biology [52]. Several studies have shown that a high rate of telomere attrition is associated with an elevated risk of coronary artery disease, myocardial infarction (MI) and heart failure [5261]. For example, in the West of Scotland Primary Prevention study (WOSCOPS) mean telomere length of peripheral blood leukocytes (LTL) was shorter in patients with severe triple vessel coronary artery disease than in individuals with angiographically normal coronary arteries [58] . In addition, associations between a reduced telomere length and the severity of CVD have been reported [56]. Cardiovascular risk factors like hypertension seem also to be related to telomere biology. In fact, both reduced telomere length and telomere uncapping were found in patients with hypertension [59, 60] [ So far only two prospective studies have been published. In both studies telomere length was an independent predictor of MI and stroke [62, 63].
T2DM is another important cardiovascular risk factor and early evidence suggests that altered telomere biology may contribute to the development of the disease [64-70]. A recent meta-analysis performed on nine cohorts with a total of 5759 cases and 6518 controls indicated that shortened telomere length is significantly associated with T2DM risk [67]. Furthermore, telomere shortening seems not only to be associated with the incidence of T2DM but also with progression of the disease and the number of diabetic complications, such as retinopathy, nephropathy, neuropathy and peripheral vascular disease [70]. However, not all studies have been able to show a prospective relationship between telomere length and incident T2DM [65].
Alzheimer’s disease (AD) is the most common neurodegenerative disease associated with aging. The association between LTL and the incidence of AD is still debated. Shorter LTL were found in AD patients [71], but no correlation was found between AD and telomere length of cerebral cells [72]. Moreover, in a longitudinal study LTL was not associated with changes in cognitive status of AD patients after 2 years of follow-up [73].
Cancer can also be considered an age-related disease, as its risk increases with aging. The potential link between telomere length and malignancies has been extensively studied in various types of tumor tissue and peripheral blood leukocytes. As the dynamics of telomere length differs between tissue and blood cells it is important to distinguish between these two approaches [74]. Reduced telomere length and poorer survival were observed in breast and prostate cancer cells as well as in sarcoma cells [74]. These findings could be explained by the Hayflick limit: telomeres become shorter at each cell division until a critical telomere length is reached. Cells with critically short telomeres undergo senescence and/or apoptosis. However, if the check-point is bypassed, cells continue to proliferate, which leads to genomic instability, accumulation of mutations and development of malignancies [75]. Additional factors like oxidative stress and chronic inflammation can aggravate this phenomenon and accelerate tumor formation [75]. However, to date it is not clear if telomere shortening is a cause or a consequence of tumor development and further studies are needed to clarify this important aspect.
Long telomeres have been shown to be associated with worse prognosis in carcinoma of the liver, colon, esophagus, head and neck [74]. Several explanations for this finding have been proposed. For example, estrogen-dependent anti-oxidant effects could contribute to telomere maintenance in breast cancer and other hormone-related malignancies [75] . It has also been speculated that cells with longer telomeres have an increased telomerase activity. Telomerase-stimulating factors, such as interleukines 2-4-6-7-10 and 13, may induce and maintain telomerase activity in these cells [75]. The presence of longer telomeres may delay senescence so that cells with long telomeres have a prolonged life span and consequently may encounter more situations, where DNA damaging stimuli can cause genetic abnormalities and chromosomal instability that ultimately lead to a malignant transformation of the affected cell.
The majority of existing studies indicate that alterations of telomere length in tumor tissue are associated with a worse prognosis. In addition, it has been speculated that tumor etiology and the stage of tumor progression may play a pivotal role for the development of alterations in telomere length [74]. For example, in clear cell renal cell carcinoma no association between telomere length in tumor tissue and patient survival was observed [74]. In any case the predictive value of telomere length measurement in tumor tissue is largely limited by the fact that it can only be obtained once the diagnosis has been established.
Several studies have investigated the relationship between LTL and cancer risk or prognosis and results are conflicting [75]. Shorter LTL were found in different cancer types, including head/neck, lung, kidney, bladder, ovarian, breast, gastric, skin, esophagus, osteosarcoma and non-Hodgkin lymphoma [75, 76]. In contrast, numerous studies indicate that cancer risk is associated with longer LTL [75] . This was found in cancers of the skin, breast, lung, kidney, hepatocellular carcinoma and non-Hodgkin lymphoma [75]. Finally, non-associations between LTL and cancer risk have been found in breast, prostate, colon and endometrial cancers [75].
These observations suggest that the timing of sample collection is an important factor that may explain some of the discordant results in previous studies that investigated the association between LTL and cancer risk. It is also possible that LTL lengthening could be the consequence of an activation of the immune system during tumor formation [75]. An alternative explanation could be that when LTL becomes critically short, compensatory mechanisms, such as hTERT activation and alternative non-telomerase- based mechanisms that maintain telomere integrity, are switched on [75]. In addition, LTL may also be modified by cancer treatment [75]. Finally, there are differences in telomere length between subtypes of blood leukocyte that further limit the interpretation of LTL results [75]. Differences in study design, cancer type, sample processing, LTL measurement and patient characteristics may be contributing factors to contradictory results in telomere length association studies [75]. Although a number of studies have investigated the association between telomere length in tumor cells or in peripheral blood leukocytes and cancer progression or survival this relationship remains insufficiently understood and further studies are needed.
Another frequent condition of aging is osteoporosis. There are conflicting reports in the literature regarding the association of telomere shortening and age-related bone loss. In the TwinsUK cohort study, LTL was independently associated with a decrease in bone mineral density (BMD) and longer LTL was also associated with reduced risk of clinical osteoporosis [77]. In contrast, in the Health Aging and Body Composition Study (Health ABC), LTL was not associated with BMD, change in BMD over 5 years, osteoporosis or fractures at baseline or after 7 years of follow-up [78].
Although there is evidence for association between telomere length and age-related diseases, neither a conclusive causative link nor a predictable association can be established. Longitudinal studies as well as assessment of other markers of telomere biology are needed to further clarify the role of telomeres in aging and the development of age-related diseases.
Vitamin D
Metabolism and physiologic aspects of vitamin D
Vitamin D metabolism is a complex process involving the action of UV radiation and hydroxylation steps (Figure 3A). Vitamin D3 is primarily produced in the skin through the action of UV-B light on 7-dehydrocholesterol [80]. In most individuals, dietary intake, from sources such as wild-caught fatty fish, provides only a small additional contribution to total vitamin D levels [81].
Vitamin D requires two hydroxylation steps to reach its active form. The first hydroxylation, occurs in the liver and produces 25-hydroxyvitamin D. (25-OHD3), the predominant form of the vitamin found in the circulation. 25-OHD3 undergoes further hydroxylation at the C1 position in the proximal tubule of the kidney [82]. The 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] thus formed is responsible for most, if not all, of the biological actions of the vitamin. A number of extra-renal tissues have demonstrated the ability to convert 25-OHD. to 1,25(OH).D3. The 1,25(OH)2D3 produced by these tissues appears to act locally in an autocrine or paracrine fashion and does not contribute significantly to the circulating 1,25(OH)2D concentration [83] .
Genomic actions
1,25(OH)2D3 primarily exerts its effects on target tissues through genomic actions via the vitamin D receptor (VDR) (Figure 3C). After binding 1,25(OH)2 D3, VDR forms heterodimers with one of three retinoid X receptors (RXRa, RXRp, RXRy) The VDR-RXR heterodimers interact with specific enhancer elements in DNA, known as vitamin D response elements (VDREs), as well as co-activators, and induce gene transcription [84, 85]. The VDR-RXR complex may also suppress the expression of certain genes, such as those encoding PTH or CYP27B1 [86, 87]. This may be mediated by the specific VDRE causing the VDR to interact with co-repressor molecules, via epigenetic mechanisms or by the modulation of specific microRNAs [88, 89]. There are thousands of VDREs, which are responsible for the cell-specific regulation of more than 200 hundred genes [5, 90] .
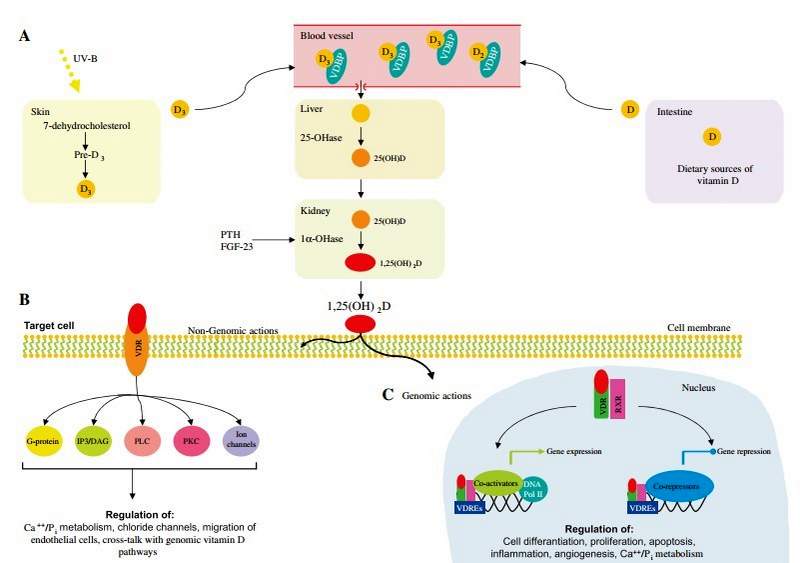
Figure 3: Vitamin D metabolism.
(A) Vitamin D3 is produced in the skin through the action of UV-B light on 7-dehydrocholesterol. A small amount of vitamin D can also be obtained from dietary sources, such as wild-caught fatty fish, mushrooms and milk. Circulating vitamin D is bound to Vitamin D Binding Protein (VDBP). In the liver vitamin D is hydroxylated in 25-hydroxy-vitamin D [25(OH)D]. Subsequently, 25(OH)D is hydroxylated in the bioactive 1a,25-dihydroxy-vitamin D [1,25(OH)2D] by the enzyme 1a-hydroxylase (1a-OHase), predominantly in the kidney. 1,25(OH)2D exerts its biological functions via non-genomic (B) and genomic (C) actions. DNA Pol II, DNA polymerase II; IP3/DAG, inositol triphosphate/dia- cylglycerol; PKC, protein kinase C; PLC, phospholipase C; RXR, retinoid X receptors; VDR, vitamin D receptor; VDREs, vitamin D response elements. Modified from Deeb et al. [79].
The vitamin D receptor has been located in many cell types, including enterocytes, myocytes, immune cells (including activated T and B lymphocytes, macrophages and dendritic cells) as well as neurons and glial cells of the central nervous system, among many others [5, 91, 92]. Vitamin D therefore exerts numerous genomic actions, including control of calcium and phosphate exchange across the cell membranes of enterocytes, renal tubular cells and skeletal muscle myocytes [92, 93]. Such genomic pathways facilitate calcium absorption from the gut, renal reabsorption of calcium and normal skeletal muscle function [5] .
Vitamin D is also involved in immune system regulation via genomic actions. It causes up-regulation of cytokine signals, antimicrobial peptides and the development of regulatory T cells [94]. Vitamin D also appears to exert genomic signals regulating cell proliferation, differentiation and apoptosis [95], while also inhibiting angiogenesis [96]. 1,25(OH)2D3 also appears to have a role in the central nervous system in detoxification, protection against free radicals, neuronal calcium regulation, immunomodulation and enhanced nerve conduction [97]. In the cardiovascular system, vitamin D down-regulates renin transcription [98] and also prevents impairments in cardiac relaxation and contractility [99] and the development of left ventricular hypertrophy [100]. The role of the VDR in telomere biology is discussed in the section ‘Vitamin D and telomere biology’.
Non-genomic actions
There are a number of responses produced by 1,25(OH)2D3 that have been shown to occur in a time-frame too rapidto be mediated by gene transcription (Figure 3B). Furthermore, these actions are not impaired by the presence of inhibitors of gene transcription or translation. It appears that these responses are mediated by the VDR located within cell membranes, in specific locations important in signal transduction pathways, known as caveolae [ 101]. When the VDR is associated with caveolae, binding of 1,25(OH)2D3 may activate a variety of second messengers, including G protein-couple receptors, inositol triphos- phate/diacylglycerol (IP3/DAG), phospholipase C (PLC), protein kinase C (PKC) or the opening of ion channels [102].
1,25-OH2D has been found to have a number of effects on transcellular calcium fluxes via non-genomic pathways. This includes the enhanced intestinal absorption of calcium as well as increases in intracellular calcium concentrations in skeletal and cardiac myocytes, fibroblasts and osteoblasts [103-107]. Non-genomic effects 1,25(OH)2D3 have also been demonstrated on calcium signaling in pancreatic p-cell cells [108], the rate of migration of endothelial cells [109], the opening of calcium and chloride channels in Sertoli cells [110] as well as increases in second messenger systems in parathyroid cells, osteoblasts, hepatocytes, chondrocytes and epithelial cells [106, 111, 112]. There appear to be instances of cross-talk between the rapid response and genomic vitamin D pathways. For example, 1,25(OH)2D3 acting via a non-genomic pathway on rat osteosarcoma cells was found to cause an increase in osteocalcin gene transcription [113].
Vitamin D deficiency and related diseases
The classical role of vitamin D is in the maintenance of adequate calcium and phosphate status. Severe vitamin D deficiency causes rickets and osteomalacia, while more moderate deficiency is associated with osteoporosis and increased fracture risk [114]. The discovery that VDR exists in multiple tissues unrelated to vitamin D’s classical function, has led to intense interest in the role of vitamin D in diverse aspects of health. Epidemiological data support an association between vitamin D deficiency and numerous conditions. These include various parameters of muscle function, multiple autoimmune diseases, upper respiratory tract infection (URTI), tuberculosis, insulin resistance, T2DM, coronary heart disease, heart failure and peripheral vascular disease and all-cause mortality [US- 121]. Observational data suggest an association between hypovitaminosis D and cognitive function, depression, bipolar disorder and schizophrenia [ 122]. Furthermore, there is an association between vitamin D and a number of cancers, including colorectal and prostate cancer [123].
Despite these associations, there are limited data from randomized controlled trials demonstrating that supplementing deficient individuals with vitamin D improves clinical outcomes. However, there is evidence that supplementation improves muscle function, prevents type 1 diabetes and multiple sclerosis, as well as diminishes clinical exacerbations in those with pre-existing multiple sclerosis [124-126]. Supplementation studies have also shown accelerated clinical recovery from tuberculosis, improvement in depressive symptoms and, in males supplemented during the first year of life, a decreased risk of schizophrenia [127-129]. Results from studies addressing domains, such as mortality, and diabetes risk, URTI and cancer have been mixed [130 -133] .
Vitamin D and aging
Mounting evidence links deficiencies of micronutrients, such as vitamin D, folate and vitamin B12 to a series of age- related diseases including neurodegenerative diseases, hypertension, CVD, T2DM and osteoporosis. Deficiencies of these vitamins are common in elderly individuals. Vitamin D deficiency, for example, affects up to 100% of elderly subjects, especially if they are house-bound or live in nursing homes [134]. Considering the role of these vitamins in cell viability, DNA synthesis and repair it can be speculated that deficiencies accelerate telomere shortening and lead to genomic instability.
Vitamin D and senescence
The role of vitamin D in cellular aging and senescence is the consequence of its numerous functions in the regulation of cellular proliferation, differentiation and apoptosis, as illustrated in Figure 4. Vitamin D regulates a range of proteins that are involved in the cell cycle, such as cyclines, cyclin-dependent kinases (CDKs) and the cyclin- dependent kinase inhibitor (CDKIs) p21 and p27. All these proteins are involved in the G1/S phase transition [79]. Once activated, these CDKIs inactivate cyclins D1, 2, 3 and E that also lose their capacity to phosphorylate pRB. Hypo- phosphorylation of pRB leads the G0/G1 cell cycle arrest and inhibition of proliferation [79]. CDKIs act as negative regulators of cell growth, as they cause G1 arrest. Several genes, including p15, p18, p21 and p27 have also been found to be regulated by vitamin D. For example, a functional VDRE was found in the promoter of the p21 coding gene, suggesting a direct regulation of p21 expression by Mineral metabolism Apoptosis
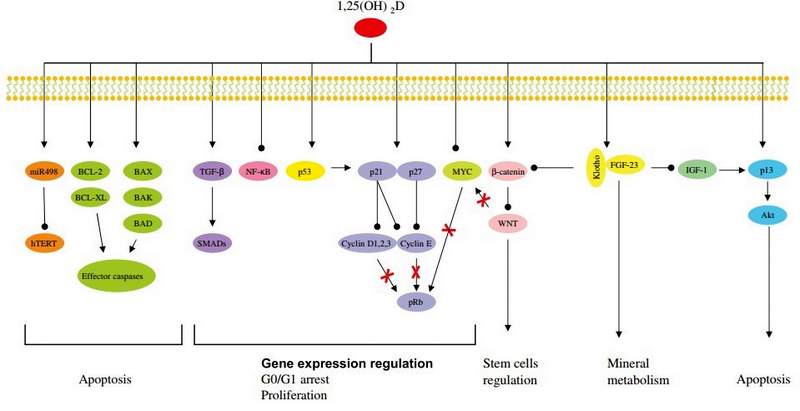
Figure 4: Vitamin D involvement in cellular aging and telomere biology.
Vitamin D influences several pathways involved in the regulation of cell growth, proliferation (TGF-p, NF-kB, p53, p21, p27 and MYC), apoptosis (hTERT, BCL-2, BCL-XL, BAX, BAK, BAD and p13), stem cell regulation (Wnt), mineral metabolism (Klotho-FGF-23). Modified from Deeb et al. [79].
VDR [135]. CDK activity can also be enhanced by the protooncogene c-myc via functional inactivation of p21 and p27. C-myc has been found to be down-regulated by vitamin D in several cell lines, leading also to cell cycle arrest [135]. In addition, vitamin D can have many indirect effects on cell-cycle regulation, due to its cross-talk with other pathways. For example, vitamin D can up-regulate the expression of the transforming growth factor p (TGF-P) signaling cascade and down-regulate the epidermial growth factor receptor (EGFR) signaling pathway [79]. In addition, recently, Strambolsky et al. have identified that vitamin D is able to enhance the physical interaction of VDR with p53 [ 136] , a well known cell cycle regulator. Vitamin D seems also to inhibit the NF-kB pathway in human leukocytes, through the down-regulation of the transcription of c-Rel and p50, two essential subunits of NF-kB [137] , The transcription factor NF-kB is a central component of the cellular response to damage, stress or inflammation [137]. Its chronic activation is observed during aging and in numerous age-related disorders, like T2DM, CVD, osteoporosis [79].
Although the exact mechanism by which vitamin D induces apoptosis is not completely understood, there is evidence that vitamin D controls members of the BCL-2 family [ 135], This protein family consists of both anti-apoptotic proteins like BCL-2 and BCL-XL and pro- apoptotic proteins like BAX, BAK and BAD. These proteins in turn form homo- or heterocomplexes and control apoptosis by regulating mitochondrial permeability and the release of cytochrome c, which ultimately leads to the activation of the caspase cascade [135]. Vitamin D exerts also apoptotic functions, as it represses the transcription of anti-apoptotic proteins (BCL-2 and BCL-XL) or inducing the expression of pro-apoptotic proteins (BAX, BAK, BAD) [79]. For example, the down regulation of the anti- apoptotic BCL-2 protein by vitamin D is a well documented phenomenon in several cell lines [135].
Another important function of vitamin D in cellular aging is mediated by the FGF-23-Klotho axis [14]. In fact, defects of FGF-23 or Klotho lead to premature aging phenotypes. Vitamin D through the interaction with VDR induces FGF-23 expression. FGF-23 requires the co-receptor Klotho to activate the Fibroblast Growth Factor Receptor (FGFR) [13]. This interaction leads to the suppression of phosphate reabsorption and vitamin D biosynthesis in the kidney. The mechanisms by which alterations in the mineral ion homeostasis influence aging process are still not clear. The FGF-23-Klotho axis may inhibit insulin-like growth factor 1 (IGF-1) signaling pathway. IGF-1 inhibition is one of the evolutionarily conserved mechanismsfor aging suppression. Evidence of an involvement of the Klotho-FGF-23 axis in the IGF-1 signaling pathway comes from mouse models. Both, Klotho- and FGF-23-deficient mice are characterized by hypoglycemia and extreme sensitivity to insulin [ 13]. In contrast, transgenic mice overexpressing Klotho are moderate resistant to insulin and IGF-1, without overt diabetes and are long-lived [13, 138]. The mechanism by which secreted Klotho suppresses insulin/IGF-1 is still unknown [ 138]. Moreover, it is recently reported that Klotho inhibits Wnt signaling pathway [13]. Wnt is essential for stem cell proliferation, in fact, chronic stimulation of Wnt signaling can lead to a rapid exhaustion of stem cells. Stem cell dysfunction contributes to aging processes and the capacity of Klotho to inhibit Wnt signaling could explain the anti-aging effect of Klotho [13]. Finally, Klotho protects cells from oxidative stress, probably regulating nitric oxide (NO) production or superoxide dismutase 2 (SOD) ex
1. Vitamin D and telomere biology
1. Studies in humans
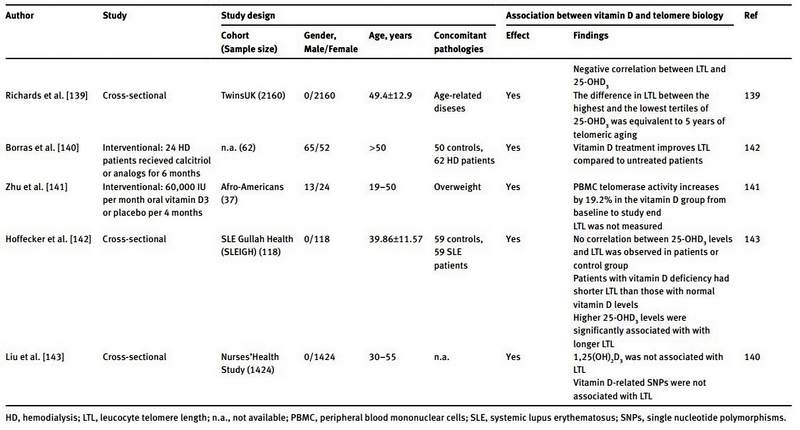
A summary of the literature data regarding the link between telomere biology and vitamin D is reported in Table 1. Richards et al. were the first to demonstrate a positive correlation between serum 25-OHD and LTL in humans, which remained significant after adjustment for age [139]. In their study they analyzed 2160 women of the TwinsUK cohort. After multiple adjustments for age, season of vitamin D measurement, menopausal status, use of hormone replacement therapy and physical activity, the difference in telomere length between the highest (124±37.3 nmol/L) and lowest (40.9±11 nmol/L) tertile of 25-OHD concentrations was equivalent to 5 years of telomeric aging [139]. In addition, the authors analyzed a subpopulation of 700 women that used vitamin D supplements. On average, these women had longer telomeres than women who did not use vitamin D supplements. However this difference was not statistically significant. These initial findings were confirmed by a subsequent study performed by Liu et al. [143]. In this study, analyses were performed on 1424 women of the Nurses’ Health Study and the results showed a positive correlation between LTL and serum 25-OHD concentrations. Logistic regression analysis indicated a concentration-dependent relationship. However, calcium intake modified this association significantly. 1,25(OH)2D was also measured, but did not correlate with LTL. As single nucleotide polymorphisms (SNPs) in genes involved in vitamin D metabolism (like SNPs in VDR, VDBP CYP2R1 and DHCR7), are reported to affect vitamin D blood concentrations, Liu et al. analyzed vitamin D-related SNPs in their population. They identified a positive association between rs7041 and rs4588 (both SNPs of VDBP), and 25-OHD levels. However, none of the investigated vitamin D-related SNPs were significantly associated with LTL [143]. Beside these cross-sectional studies, only one interventional study has been performed in order to clarify the effect of vitamin D supplementation on telomere biology [141]. Zhu et al. treated 37 obese Afro-American subjects in a double-blind randomized fashion with either a monthly oral dose of 60,000 IU of vitamin D3 or placebo for a period of 4 months. At the end of the study the serum 25-OHD concentration in vitamin D treated subjects was markedly increased when compared to baseline. The rise in serum 25-OHD was accompanied by a 19.2% increase of peripheral blood mononuclear cell (PBMC) telomerase activity. In the placebo group no such changes were seen. However, LTL was not measured in this study, limiting also the power of their findings. Further support for vitamin supplementation preserving telomere length comes from a recent study in hemodialysis patients [140]. Borras et al. observed longer telomeres in hemodialysis patients treated with calcitriol or analogs for at least 6 months compared to hemodialysis patients without such treatment [140]. Finally, Hoffecker et al. reported a significant correlation between telomere length and serum 25-OHD concentrations in vitamin D deficient (serum 25-OHD <20 ng/mL) patients with systemic lupus erythematosus (SLE) and unaffected controls [142]. Moreover, patients with SLE whose serum 25-OHD concentrations remained insufficient/deficient (<30 ng/mL) after 2.8 years of follow-up had shorter telomeres than patients with a sufficient serum 25-OHD concentration (>30 ng/mL).
1. Animal studies
So far, animal models have not been used to study the role of vitamin D in telomere biology. However, a limited number of animal studies have investigated the involvement of vitamin D in cellular aging and the results mainly support a significant relationship. VDR knockout mice (VDR -/-), e.g., have been used to study the function of vitamin D in cellular aging processes [144]. Keisala et al. have shown that these mice develop signs of premature aging, such as infertility, muscle atrophy, reduced immune function, and osteoporosis [ 145]. Furthermore, VDR -/- mice have a shorter life span [145]. The phenotype of premature aging in these animals was accompanied by a reduced expression of NF-kB, FGF-23 and p53 [144, 145]. An alteration of p53 and NF-kB expression by vitamin D is also supported by a two other studies [79, 136].
These pathways are known to play an important role in cellular senescence [8]. Valcheva et al. have also shown that in VDR -/- mice vascular smooth muscle cells produced higher intracellular superoxide anion and thus promoting premature senescence [146].
Beside the VDR -/- mice model, other two mice models (FGF-23 -/- and Klotho -/-) have been utilized for studying the relationship of vitamin D pathway and aging. In fact, both FGF-23 and Klotho deficient mice have increased renal expression of e 1a-hydroxylase, accompanied by significantly elevated serum levels of 1,25(OH)2D, infertility, atherosclerosis, skin atrophy, muscle wasting, T-cell deregulation and short lifespan [144]. A significant rescue of these phenotypes has been achieved providing a vitamin D deficient diet [144]. The hypothesis that hypo- or hyper-vitaminosis D causes accelerated aging is supported by animal studies that show a U-shaped association between serum 25(OH)2D and the risk of cancer [144]. Based on these results it can also be speculated that there is an optimal serum concentration for general health [144].
1. In vitro studies
Further evidence for a role of vitamin D in telomere biology and cellular aging comes from cell culture studies. Treatment of different cell lines with vitamin D drives cell differentiation and has been shown to dramatically reduce telomerase activity [147]. For example, a reduction of telomerase activity was observed after treating leukemic HL-60 cells for 5 or 7 days with 100 ng/mL of vitamin D3 [147]. However, the significance of this study is limited by the fact that no other vitamin D concentration was tested. Therefore, it remains unclear if there is a concentration- dependent relationship between telomerase activity and vitamin D. A similar phenomenon was observed in human prostate cancer cells [148]. Treatment of these cells with a combination of 1,25(OH)2D and 9-cis-retinoic acid significantly reduced telomerase activity. The authors hypothesized a direct interaction of the VDR/RXR heterodimer with the DR3 ' sequence in the human telomerase reverse transcriptase (hTERT) promoter as the underlying mechanism. The DR3'sequence (5'-AGTTCATGGAGTTCA-3') is a vitamin D response element present in the promoter region of the hTERT gene [148]. Jiang et al. have described an alternative mechanism in an ovarian cancer cell line. Vitamin D downregulates hTERT activity in these cells by decreasing the stability of hTERT mRNA [149]. Kasiappan et al. proposed another appealing mechanism of vitamin D dependent telomerase regulation through small noncoding RNA molecules. They were able to demonstrate that vitamin D can induce microRNA-498 (miR-498) ex
However, all these studies were performed on cancer cell lines that are characterized by genomic instability, chromosome alterations and other features that are associated with transformed cells. Therefore it is not possible to extrapolate the real role of vitamin D in telomere biology in healthy normal cells. Further studies are needed to clarify the role of vitamin D in telomere biology.
- Conclusions
In summary, existing evidence supports the concept that vitamin D contributes to cellular aging and telomere biology. For example, human studies show an inverse relationship between serum vitamin D and age-related diseases as well as serum vitamin D and mortality. At a cellular level, vitamin D appears to regulate proliferation, senescence and apoptosis, through genomic and nongenomic pathways. One pathway through which vitamin D can delay cellular aging is the preservation of telomere biology. However, all studies published so far harbor significant limitations and results are sometimes conflicting. Human studies are predominantly of cross-sectional nature and the few longitudinal studies have been too small to draw conclusive findings. Animal and cell culture studies are very heterogeneous as they have used different animal models and cell types. This hampers the comparison of results and makes general conclusions impossible. Further systematic studies are needed to understand the role of vitamin D in telomere biology and cellular aging. In particular, large prospective studies will help to clarify if vitamin D supplementation can delay aging and reduce the risk of developing age-related diseases.
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
Financial support: None declared.
Employment or leadership: None declared.
Honorarium: None declared.
Competing interests: The funding organization(s) played no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the report for publication.
1. References
(Note numbers may not agree with the text)
Muller HJ. Bar Duplication. Science 1936;83:528-30.
Varela E, Blasco MA. 2009 Nobel prize in physiology or medicine: telomeres and telomerase. Oncogene 2010;29:1561-5.
Armanios M. Telomeres and age-related disease: how telomere biology informs clinical paradigms. J Clin Invest 2013;123:996 - 1002.
Paul L. Diet, nutrition and telomere length. J Nutr Biochem 2011;22:895 - 901.
Holick MF. Vitamin D deficiency. N Engl J Med 2007;357:266-81.
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013;153:1194-217.
Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013;123:966-72.
Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol 2013;75:685 - 705.
Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009;461:1071-8.
Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep 2014;15:1139-53.
Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis 2008;4:68-75.
Russell SJ, Kahn CR. Endocrine regulation of ageing. Nat Rev Mol Cell Biol 2007;8:681 - 91.
Kuro-o M. Klotho as a regulator of oxidative stress and senescence. Biol Chem 2008;389:233-41.
Lanske B, Razzaque MS. Mineral metabolism and aging: the fibroblast growth factor 23 enigma. Curr Opin Nephrol Hyper- tens 2007;16:311-8.
Kuro-o M. Klotho, phosphate and FGF-23 in ageing and disturbed mineral metabolism. Nat Rev Nephrol 2013;9:650-60.
Blackburn EH. Telomeres and telomerase: the means to the end (Nobel lecture). Angew Chem Int Ed Engl 2010;49:7405-21.
Blackburn EH. Switching and signaling at the telomere. Cell 2001;106:661 - 73.
Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 2005;6:611-22.
Blasco MA. Telomere length, stem cells and aging. Nat Chem Biol 2007;3:640 - 9.
Blasco MA. Mice with bad ends: mouse models for the study of telomeres and telomerase in cancer and aging. EMBO J 2005;24:1095 - 103.
Raynaud CM, Sabatier L, Philipot O, Olaussen KA, Soria JC. Telomere length, telomeric proteins and genomic instability during the multistep carcinogenic process. Crit Rev Oncol Hema- tol 2008;66:99-117.
Xin H, Liu D, Songyang Z. The telosome/shelterin complex and its functions. Genome Biol 2008;9:232.
O'Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 2010;11:171 - 81.
Smogorzewska A, de Lange T. Regulation of telomerase by telomeric proteins. Annu Rev Biochem 2004;73:177-208.
Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 1965;37:614-36.
Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science 1994;266:2011-5.
Cukusic A, Skrobot VN, Sopta M, Rubelj I. Telomerase regulation at the crossroads of cell fate. Cytogenet Genome Res 2008;122:263-72.
Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985;43:405 - 13.
Marrone A, Dokal I. Dyskeratosis congenita: molecular insights into telomerase function, ageing and cancer. Expert Rev Mol Med 2004;6:1 - 23.
Baird DM. New developments in telomere length analysis. Exp Gerontol 2005;40:363 - 8.
Flores I, Canela A, Vera E, Tejera A, Cotsarelis G, Blasco MA. The longest telomeres: a general signature of adult stem cell compartments. Genes Dev 2008;22:654-67.
Canela A, Vera E, Klatt P, Blasco MA. High-throughput telomere length quantification by FISH and its application to human population studies. Proc Natl Acad Sci USA 2007;104:5300-5.
Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res 2002;30:2-6.
Cawthon RM. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res 2009;37:1-7.
O'Callaghan NJ, Fenech M. A quantitative PCR method for measuring absolute telomere length. Biol Proced Online 2011;13:1 - 13.
Martin-Ruiz CM, Baired D, Roger L, Boukamp P, Krunic D, Cawthon R, et al. Reproducibility of telomere length assessment: an international collaborative study. Int J Epidemiol 2014;1-11.
Mathon NF, Lloyd AC. Cell senescence and cancer. Nat Rev Cancer 2001;1:203-13.
Batista LF, Artandi SE. Telomere uncapping, chromosomes, and carcinomas. Cancer Cell 2009;15:455-7.
Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet 2012;13:693-704.
Fyhrquist F, Saijonmaa O, Strandberg T. The roles of senescence and telomere shortening in cardiovascular disease. Nat Rev Cardiol 2013;10:274 - 83.
Mazumdar R. Telomere length and aging. Biochemistry, genetics and molecular biology. Rev Select Topics Telomere Biol 2012:3 - 30.
Cawthon RM, Smith KR, O'Brien E, Sivatchenko A, Kerber RA. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 2003;361:393-5.
Bakaysa SL, Mucci LA, Slagboom PE, Boomsma DI, McClearn GE, Johansson B, et al. Telomere length predicts survival independent of genetic influences. Aging Cell 2007;6:769-74.
Ehrlenbach S, Willeit P, Kiechl S, Willeit J, Reindl M, Schanda K, et al. Influences on the reduction of relative telomere length over 10 years in the population-based Bruneck Study: introduction of a well-controlled high-throughput assay. Int J Epidemiol 2009;38:1725-34.
Fitzpatrick AL, Kronmal RA, Kimura M, Gardner JP, Psaty BM, Jenny NS, et al. Leukocyte telomere length and mortality in the Cardiovascular Health Study. J Gerontol A Biol Sci Med Sci 2011;66:421 - 9.
Kimura M, Hjelmborg JV, Gardner JP, Bathum L, Brimacombe M, Lu X, et al. Telomere length and mortality: a study of leukocytes in elderly Danish twins. Am J Epidemiol 2008;167:799-806.
Bendix L, Thinggaard M, Fenger M, Kolvraa S, Avlund K, Linneberg A, et al. Longitudinal changes in leukocyte telomere length and mortality in humans. J Gerontol A Biol Sci Med Sci 2014;69:231 - 9.
Bischoff C, Petersen HC, Graakjaer J, Andersen-Ranberg K, Vaupel JW, Bohr VA, et al. No association between telomere length and survival among the elderly and oldest old. Epidemiology 2006;17:190-4.
Fitzpatrick AL, Kronmal RA, Gardner JP, Psaty BM, Jenny NS,
Tracy RP, et al. Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am J Epidemiol 2007;165:14 - 21.
Harris SE, Deary IJ, MacIntyre A, Lamb KJ, Radhakrishnan K,
Starr JM, et al. The association between telomere length, physical health, cognitive ageing, and mortality in non-demented older people. Neurosci Lett 2006;406:260-4.
Svensson J, Karlsson MK, Ljunggren O, Tivesten A, Mellstrom D, Moverare-Skrtic S. Leukocyte telomere length is not associated with mortality in older men. Exp Gerontol 2014;57:6-12.
Nilsson PM, Tufvesson H, Leosdottir M, Melander O. Telomeres and cardiovascular disease risk: an update 2013. Transl Res 2013;162:371 - 80.
Brouilette S, Singh RK, Thompson JR, Goodall AH, Samani NJ. White cell telomere length and risk of premature myocardial infarction. Arterioscler Thromb Vasc Biol 2003;23:842-6.
Benetos A, Gardner JP, Zureik M, Labat C, Xiaobin L, Adamo- poulos C, et al. Short telomeres are associated with increased carotid atherosclerosis in hypertensive subjects. Hypertension 2004;43:182 - 5.
Willeit P, Willeit J, Brandstatter A, Ehrlenbach S, Mayr A,
Gasperi A, et al. Cellular aging reflected by leukocyte telomere length predicts advanced atherosclerosis and cardiovascular disease risk. Arterioscler Thromb Vasc Biol 2010;30:1649-56.
van der Harst P, van der Steege G, de Boer RA, Voors AA, Hall AS, Mulder MJ, et al. Telomere length of circulating leukocytes is decreased in patients with chronic heart failure. J Am Coll Cardiol 2007;49:1459 - 64.
Samani NJ, Boultby R, Butler R, Thompson JR, Goodall AH. Telomere shortening in atherosclerosis. Lancet 2001;358: 472 - 3.
Brouilette SW, Moore JS, McMahon AD, Thompson JR, Ford I, Shepherd J, et al. Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case-control study. Lancet 2007;369:107 - 14.
Jeanclos E, Schork NJ, Kyvik KO, Kimura M, Skurnick JH, Aviv A. Telomere length inversely correlates with pulse pressure and is highly familial. Hypertension 2000;36:195-200.
Morgan RG, Ives SJ, Walker AE, Cawthon RM, Andtbacka RH, Noyes D, et al. Role of arterial telomere dysfunction in hypertension: relative contributions of telomere shortening and telomere uncapping. J Hypertens 2014;32:1293-9.
Hoffmann J, Spyridopoulos I. Telomere length in cardiovascular disease: new challenges in measuring this marker of cardiovascular aging. Future Cardiol 2011;7:789-803.
Weischer M, Bojesen SE, Cawthon RM, Freiberg JJ, Tybjaerg- Hansen A, Nordestgaard BG. Short telomere length, myocardial infarction, ischemic heart disease, and early death. Arterioscler Thromb Vasc Biol 2012;32:822-9.
Perez-Rivera JA, Pabon-Osuna P, Cieza-Borrella C, Martin- Herrero F, Gonzalez-Porras JR, Gonzalez-Sarmiento R. Prognostic value of telomere length in acute coronary syndrome. Mech Ageing Dev 2012;133:695-7.
Gardner JP, Li S, Srinivasan SR, Chen W, Kimura M, Lu X, et al. Rise in insulin resistance is associated with escalated telomere attrition. Circulation 2005;111:2171-7.
You NC, Chen BH, Song Y, Lu X, Chen Y, Manson JE,et al.A prospective study of leukocyte telomere length and risk of type 2 diabetes in postmenopausal women. Diabetes 2012;61: 2998 - 3004.
Zhao J, Zhu Y, Lin J, Matsuguchi T, Blackburn E, ZhangY, et al. Short leukocyte telomere length predicts risk of diabetes in American Indians: the strong heart family study. Diabetes 2014;63:354 - 62.
Zhao J, Miao K, Wang H, Ding H, Wang DW. Association between telomere length and type 2 diabetes mellitus: a meta-analysis. PLoS One 2013;8:e79993.
Elks CE, Scott RA. The long and short of telomere length and diabetes. Diabetes 2014;63:65 - 7.
Olivieri F, Lorenzi M, Antonicelli R, Testa R, Sirolla C, Cardelli M, et al. Leukocyte telomere shortening in elderly Type2DM patients with previous myocardial infarction. Atherosclerosis 2009;206:588 - 93.
Testa R, Olivieri F, Sirolla C, Spazzafumo L, Rippo MR, Marra M, et al. Leukocyte telomere length is associated with complications of type 2 diabetes mellitus. Diabet Med 2011;28:1388-94.
Thomas P, O' Callaghan NJ, Fenech M. Telomere length in white blood cells, buccal cells and brain tissue and its variation with ageing and Alzheimer's disease. Mech Ageing Dev 2008;129:183 - 90.
Lukens JN, Van Deerlin V, Clark CM, Xie SX, Johnson FB. Comparisons of telomere lengths in peripheral blood and cerebellum in Alzheimer's disease. Alzheimers Dement 2009;5:463-9.
Zekry D, Herrmann FR, Irminger-Finger I, Ortolan L, Genet C, Vitale AM, et al. Telomere length is not predictive of dementia or MCI conversion in the oldest old. Neurobiol Aging 2010;31:719 - 20.
Svenson U, Roos G. Telomere length as a biological marker in malignancy. Biochim Biophys Acta 2009;1792:317-23.
Hou L, Zhang X, Gawron AJ, Liu J. Surrogate tissue telomere length and cancer risk: shorter or longer? Cancer Lett 2012;319:130 - 5.
Wu X, Amos CI, Zhu Y, Zhao H, Grossman BH, Shay JW, et al. Telomere dysfunction: a potential cancer predisposition factor. J Natl Cancer Inst 2003;95:1211-8.
Valdes AM, Richards JB, Gardner JP, Swaminathan R, Kimura M, Xiaobin L, et al. Telomere length in leukocytes correlates with bone mineral density and is shorter in women with osteoporosis. Osteoporos Int 2007;18:1203-10.
Sanders JL, Cauley JA, Boudreau RM, Zmuda JM, Strotmeyer ES, Opresko PL, et al. Leukocyte telomere length is not associated with BMD, osteoporosis, or fracture in older adults: results from the health, aging and body composition study. J Bone Miner Res 2009;24:1531-6.
Deeb KK, Trump DL, Johnson CS. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer 2007;7:684 - 700.
Holick M, MacLaughlin J, Clark M, Holick S. Photosynthesis of previtamin D3 in human skin and the physiologic consequences. Science 2010;210:203-5.
National Institute of Health. Dietary supplement fact sheet: vitamin D. Available from: http://ods.od.nih.gov/factsheets/ VitaminD-HealthProfessional/. Accessed on 3 June 2014.
Lehmann B. The vitamin D3 pathway in human skin and its role for regulation of biological processes. Photochem Photobiol 2005;81:1246 - 51.
Prentice A, Goldberg GR, Schoenmakers I. Vitamin D across the lifecycle: physiology and biomarkers. Am J Clin Nutr 2008;88:500S - 6S.
Kato S. The function of vitamin D receptor in vitamin D action. J Biochem 2000;127:717-22.
Freedman LP. Increasingthecomplexity of coactivation in nuclear receptor signaling. Cell 1999;97:5-8.
Amizuka N, Kwan MY, Goltzman D, Ozawa H, White JH. Vitamin D3 differentially regulates parathyroid hormone/parathyroid hormone-related peptide receptor expression in bone and cartilage. J Clin Invest 1999;103:373-81.
Turunen MM, Dunlop TW, Carlbery C, Vaisanen S. Selective use of multiple vitamin D response elements underlies the 1alpha,25-dihydroxyvitamin D3-mediated negative response elements of the human CYP27B1 gene. Nucleic Acids Res 2007;35:2734-47.
Kim MS, Kondo T, Takada I, Youn MY, Yamamoto Y, Takahashi S, et al. DNA demethylation in hormone-induced transcriptional derepression. Nature 2009;461:1007-12.
Komagata S, Nakajima M, Takagi S, Mohri T, Taniya T, Yokoi T. Human CYP24 catalyzing the inactivation of calcitriol is post-transcriptionally regulated by miR-125b. Mol Pharmacol 2009;76:702 - 9.
Bikle DD. Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol 2014;21:319-29.
Jones G, Strugnell SA, DeLuca HF. Current understanding of the molecular actions of vitamin D. Physiol Rev 1998;78:1193-231.
Bouillon R, Carmeliet G, Verlinden L, van EE, Verstuyf A, Luderer HF, et al. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev 2008;29:726-76.
Li YC, Bolt MJ, Cao LP, Sitrin MD. Effects of vitamin D receptor inactivation on the expression of calbindins and calcium metabolism. Am J Physiol Endocrinol Metab 2001;281:E558-64.
Kamen DL, Tangpricha V. Vitamin D and molecular actions on the immune system: modulation of innate and autoimmunity. J Mol Med (Berl) 2010;88:441-50.
Lappe JM, Travers-Gustafson D, Davies KM, Recker RR, Heaney RP. Vitamin D and calcium supplementation reduces cancer risk: results of a randomized trial. Am J Clin Nutr 2007;85:1586-91.
Majewski S, Skopinska M, Marczak M, Szmurlo A, Bollag W, Jablonska S. Vitamin D3 is a potent inhibitor of tumor cell-induced angiogenesis. J Investig Dermatol Symp Proc 1996;1:97 - 101.
Buell JS, Dawson-Hughes B. Vitamin D and neurocognitive dysfunction: preventing “D”ecline? Mol Aspects Med 2008;29:415 - 22.
Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxy- vitamin D(3) is a negative endocrine regulator of the renin- angiotensin system. J Clin Invest 2002;110:229-38.
Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008;149:558-64.
Simpson RU, Hershey SH, Nibbelink KA. Characterization of heart size and blood pressure in the vitamin D receptor knockout mouse. J Steroid Biochem Mol Biol 2007;103:521-4.
Huhtakangas JA, Olivera CJ, Bishop JE, Zanello LP, Norman AW. The vitamin D receptor is present in caveolae-enriched plasma membranes and binds 1 alpha,25(OH)2-vitamin D3 in vivo and in vitro. Mol Endocrinol 2004;18:2660-71.
Haussler MR, Jurutka PW, Mizwicki M, Norman AW. Vitamin D receptor (VDR)-mediated actions of 1alpha,25(OH)(2)vitamin D(3): genomic and non-genomic mechanisms. Best Pract Res Clin Endocrinol Metab 2011;25:543-59.
de Boland AR, Morelli S, Boland R. 1,25(OH)2-vitamin D3 signal transduction in chick myoblasts involves phosphatidylcholine hydrolysis. J Biol Chem 1994;269:8675-9.
Selles J, Boland R. Rapid stimulation of calcium uptake and protein phosphorylation in isolated cardiac muscle by dihydroxyvitamin D3. Mol Cell Endocrinol 1991;77:67-73.
Barsony J, Marx SJ. Rapid accumulation of cyclic GMP near activated vitamin D receptors. Proc Natl Acad Sci USA 1991;88:1436 - 40.
Lieberherr M. Effects of vitamin D3 metabolites on cytosolic free calcium in confluent mouse osteoblasts. J Biol Chem 1987;262:13168 - 73.
Le M, V, Grosse B, Lieberherr M. Phospholipase C beta and membrane action of calcitriol and estradiol. J Biol Chem 1997;272:11902-7.
Sergeev IN, Rhoten WB. 1,25-Dihydroxyvitamin D3 evokes oscillations of intracellular calcium in a pancreatic beta-cell line. Endocrinology 1995;136:2852-61.
Rebsamen MC, Sun J, Norman AW, Liao JK. 1alpha,25-dihydrox- yvitamin D3 induces vascular smooth muscle cell migration via activation of phosphatidylinositol 3-kinase. Circ Res 2002;91:17 - 24.
Menegaz D, Barrientos-Duran A, Kline A, Silva FR, Norman AW, Mizwicki MT, et al. 1alpha,25(OH)2-Vitamin D3 stimulation of secretion via chloride channel activation in Sertoli cells. J Steroid Biochem Mol Biol 2010;119:127-34.
Bourdeau A, Atmani F, Grosse B, Lieberherr M. Rapid effects of dihydroxyvitamin D3 and extracellular Ca2+ on phospholipid metabolism in dispersed porcine parathyroid cells. Endocrinology 1990;127:2738-43.
Sorensen AM, Baran DT. 1 alpha,25-Dihydroxyvitamin D3 rapidly alters phospholipid metabolism in the nuclear envelope of osteoblasts. J Cell Biochem 1995;58:15-21.
Baran D, Sorensen A, Shalhoub V, Owen T, Stein G, Lian J. The rapid nongenomic actions of 1alpha,25-dihydroxyvitamin D3
modulate the hormone-induced increments in osteocalcin gene transcription in osteoblast-like cells. J Cell Biochem 1992;50:124-9.
Lai L, Lucas R, Clements M, Roddam A, Banks E. Hip fracture risk in relation to vitamin D supplementation and serum 25-hydroxyvitamin D levels: a systematic review and metaanalysis of randomised controlled trials and observational studies. J Clin Invest 1990;85:1296-303.
Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008;102:1540-4.
Bischoff-Ferrari H, Dietrich T, Orav EJ, Hu FB, ZhangY,
Karlson EW, et al. Higher 25-hydroxyvitamin D concentrations are associated with better lower-extremity function in both active and inactive persons aged > or = 60 y. Am J Clin Nutr 2004;80:752-8.
Ginde A, Mansbach K, Camargo CJ. Association between serum 25-hydroxyvitamin D level and upper respiratory trct infection in the Third National Health and Nutrition Examination Survey. Arch Intern Med 2009;169:384-90.
Nnoaham K, Clarke A. Low serum vitamin D levels and tuberculosis: a systematic review and meta-analysis. Int J Epidemiol 2008;37:113 - 9.
Pittas A, Lau J, Dawson-Hughes B. The role of vitamin D and calcium in type 2 diabetes. J Clin Endocrinol Metab 2007;92:2017 - 9.
Schottker B, Haug U, Schomburg L, Kohrle J, Perna L, Muller H, et al. Strong associations of 25-hydroxyvitamin D concentrations with all-cause, cardiovascular, cancer, and respiratory disease mortality in a large cohort study. Am J Clin Nutr 2013;97:782 - 93.
Herrmann M, Sullivan DR, Veillard AS, McCoroquodale T,
Straub IR, Scott R, et al. Serum 25-hydroxy vitamin D: a predictor of macrovascular and microvascular complications in patients with type 2 diabetes. Diabetes Care, 2014.
Cherniack E, Troen B, Florenz H, Roos B, Levis S. Some new food for thought: the role of vitamin D in the mental health of older adults. Curr Psychiatry Rep 2009;11:12-9.
Gandini S, Boniol M, Haukka J, Byrnes G, Cox B, Sneyd MJ, et al. Meta-analysis of observational studies of serum 25-hydroxyvitamin D levels and colorectal, breast and prostate cancer and colorectal adenoma. Int J Cancer 2011;128: 1414 - 24.
Verhaar H, Samson M, Jansen P, de Vreede P, Manten J, Duursma S. Muscle strength, functional mobility and vitamin D in older women. Aging 2000;12:455-60.
Jarvelin M, Virtanen S. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet 2001;358:1500-3.
Myhr K. Vitamin D treatment in multiple sclerosis. J Neurol Sci McGrath J, Saari K, Hakko H. Vitamin D supplementation during the first year of life and risk of schizophrenia: a Finnish birth- cohort study. Schizophr Res 2004;67:237-45.
Trivedi D, Doll R, Khaw K. Effect of four monthly oral vitamin D3 (cholecalciferol) supplementation on fractures and mortality in men and women living in the community: randomized double blind controlled trial. Br Med J 2003;326:469.
Borissova A, Tankova T, Kirilov G, Dakovska L, Kovacheva R. The effect of vitamin D3 on insulin secretion and peripheral insulin sensitivity in type 2 diabetic patients. Int J Clin Pract 2003;57:258 - 61.
Li-Ng M, Aloia J, Pollack S, Cunha BA, Mikhail M, Yeh J, et al. A randomized controlled trial of vitamin D3 supplementation for the prevention of symptomatic upper respiratory tract infections. Epidemiol Infect 2009;137:1396-404.
Wactawski-Wende J, Kotchen J, Anderson G, Assaf A, Brunner R, O'Sullivan M, et al. Calcium plus vitamin D supplementation and the risk of colorectal cancer. N Engl J Med 2006;354:684 - 94.
Gloth FM, III, Gundberg CM, Hollis BW, Haddad JG Jr., Tobin JD. Vitamin D deficiency in homebound elderly persons. JAMA 1995;274:1683 - 6.
Ylikomi T, Laaksi I, Lou YR, Martikainen P, Miettinen S, Pennanen P, et al. Antiproliferative action of vitamin D. Vitam Horm 2002;64:357 - 406.
Stambolsky P, Tabach Y, Fontemaggi G, Weisz L, Maor-Aloni R, Siegfried Z, et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 2010;17: 273-85.
Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, et al. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest 2012;122:2601-12.
Kuro-o M. Klotho and the aging process. Korean J Intern Med 2011;26:113 - 22.
Richards JB, Valdes AM, Gardner JP, Paximadas D, Kimura M, Nessa A, et al. Higher serum vitamin D concentrations are associated with longer leukocyte telomere length in women. Am J Clin Nutr 2007;86:1420-5.
Borras M, Panizo S, Sarro F, Valdivielso JM, Fernandez E. Assessment of the potential role of active vitamin D treatment in telomere length: a case-control study in hemodialysis patients. Clin Ther 2012;34:849 - 56.
Zhu H, Guo D, Li K, Pedersen-White J, Stallmann-Jorgensen IS, Huang Y, et al. Increased telomerase activity and vitamin D supplementation in overweight African Americans. Int J Obes (Lond) 2012;36:805-9.
Hoffecker BM, Raffield LM, Kamen DL, NowlingTK. Systemic lupus erythematosus and vitamin D deficiency are associated with shorter telomere length among African Americans: a case- control study. PLoS One 2013;8:e63725.
Liu JJ, Prescott J, Giovannucci E, Hankinson SE, Rosner B, Han J, et al. Plasma vitamin D biomarkers and leukocyte telomere length. Am J Epidemiol 2013;177:1411-7.
Tuohimaa P. Vitamin D and aging. J Steroid Biochem Mol Biol 2009;114:78 - 84.
Keisala T, Minasyan A, Lou YR, Zou J, Kalueff AV, Pyykko I, et al. Premature aging in vitamin D receptor mutant mice. J Steroid Biochem Mol Biol 2009;115:91-7.
Valcheva P, Cardus A, Panizo S, Parisi E, Bozic M, Lopez Novoa JM, et al. Lack of vitamin D receptor causes stress-induced premature senescence in vascular smooth muscle cells through enhanced local angiotensin-II signals. Atherosclerosis 2014;235:247-55.
Sharma HW, Sokoloski JA, Perez JR, Maltese JY, Sartorelli AC, Stein CA, et al. Differentiation of immortal cells inhibits telom- erase activity. Proc Natl Acad Sci U S A 1995;92:12343-6.
Ikeda N, Uemura H, Ishiguro H, Hori M, Hosaka M, Kyo S, et al. Combination treatment with 1alpha,25-dihydroxyvitamin D3 and 9-cis-retinoic acid directly inhibits human telomerase reverse transcriptase transcription in prostate cancer cells. Mol Cancer Ther 2003;2:739-46.
Jiang F, Bao J, Li P, Nicosia SV, Bai W. Induction of ovarian cancer cell apoptosis by 1,25-dihydroxyvitamin D3 through the down- regulation of telomerase. J Biol Chem 2004;279:53213-21.
Kasiappan R, Shen Z, Tse AK, Jinwal U, Tang J, Lungchukiet P, et al. 1,25-Dihydroxyvitamin D3 suppresses telomerase expression and human cancer growth through microRNA-498. J Biol Chem 2012;287:41297 - 309.
- # 12+ VitaminDWiki pages with TELOMERE in the title
This list is automatically updated
{LIST()}