Liver and interactions with vitamin D deficiency - Review
Vitamin D in liver diseases: From mechanisms to clinical trials
Journal of Gastroenterology and Hepatology , Vol 28 Issue
- Yuan-Ping Han1,2, hanyp@scu.edu.cn, Yuan-Ping Han, Ming Kong2, Sujun Zheng2, Yan Ren2, #Longdon Zhu2, Hongbo Shi2, Zhongping Duan2
Article first published online: 15 JUL 2013DOI: 10.1111/jgh.12016
Special Issue: 7th International Symposium on Alcoholic Liver and Pancreatic Diseases and Cirrhosis.
Funding for this conference was made possible (in part) by Grant 5 R13AA20691-02 from the National Institute on Alcohol Abuse and Alcoholism (NIAAA). Guest Editors: Bin Gao and Fu-Sheng WangVolume 28, Issue Supplement S1, pages 49–55, August 2013
Funding: Supported by NIH funding (R01DK069418) and startup fund from Sichuan University to YPH, China National Key Project of the Twelfth Five-Year Plan (2012ZX10002004-006), Beijing Chaoyang District, AIDS and Viral Hepatitis Beijing (2012ZX10004904-003-001) to DZP, Beijing New Star Project on Science and Technology (2007B055) to ZSJ.
Abstract
Traditionally regarded as a typical vitamin regulating calcium and phosphorus homeostasis, vitamin D is now discovered as a highly versatile molecule with emerging roles in immunity, cancer, infectious diseases, fibrosis, fatty liver diseases, and alcoholic liver diseases. A large body of clinical evidence has demonstrated the prevalence and risks of vitamin D deficiency in various chronic diseases. Biologically active vitamin D, 1,25-dihydroxylvitamin D3, is synthesized in two distinct systems. In addition to the classic two-step hydroxylation in the liver and kidneys, 1,25-dihydroxylvitamin D3 can also be produced locally by immune cells in response to infection. The bioactive vitamin D generated in these two pools apparently functions differently: while the former facilitates calcium adsorption and homeostasis, the latter confers immune regulation. The immune regulatory functions of vitamin D are demonstrated by induction of antimicrobial peptides, suppression of innate immune response, induction of Th2 cytokines, and stimulation of T-regulatory T cells. Vitamin D deficiency or insufficiency is overwhelmingly associated with viral hepatitis, cirrhosis, and fatty liver diseases. Recent clinical trials have shown that vitamin D supplements significantly enhance the efficacy of interferon plus ribavirin therapy through sustained virological response. A recent study showed that 25-dihydroxyvitamin D rather than 1,25-dihydroxyvitamin D could directly suppress hepatitis C virus assembly. Moreover, clinical evidence has shown that vitamin D deficiency is associated with alcoholic and non-alcoholic fatty liver diseases. In this review, we highlight some recent advances in vitamin D researches and clinical trails.
Bioactive vitamin D (VD), which is synthesized by two different systems, exerts distinct functions
Different from the classical definition of a vitamin, VD is neither a co-enzyme factor nor an essential nutrient component. In addition to dietary sources from animals (VD{SUB()}3{SUB}) or plants (VD{SUB()}2{SUB}), VD can be synthesized in the skin from cholesterol under sunlight.[1] Driven by sunlight, 7-dehydrocholesterol in the skin cells is converted to pre-vitamin D3, which consequently undergoes an isomerization process to vitamin D{SUB()}3{SUB}, also known as cholecalciferol. The first step in the synthesis of biologically active VD from vitamin D{SUB()}3{SUB} occurs in hepatocytes through 25-hydroxylation, catalyzed by Cyp2R1 or Cyp27A1.[1] Secreted from hepatocytes, 25(OH)D{SUB()}3{SUB} is conveyed by VD-binding protein (VDBP) to the kidneys, where it is additionally hydroxylated by 1-alpha-hydroxylase (Cyp27B1) to generate fully activated form, 1,25-dihydroxyvitamin D, namely calcitriol. VD levels are also regulated by its degradation processes. Through a negative feedback loop, calcitriol can induce the catabolic enzyme 24-hydroxylase (Cyp24A1) in the kidneys as well as in other VD-targeting tissues, which inactivates VD and promotes it breakdown.[2] Furthermore, to ensure bioavailability, Cyp24A1 transcription is negatively regulated by parathyroid hormone (PTH) driven by low calcium levels. Serum 25(OH)D levels are usually a thousand times higher than 1,25(OH){SUB()}2{SUB}D levels, indicating that the limiting step for synthesis of active VD is conducted mostly by 1-hydroxylation via the relative activities of between its synthesis by Cyp27B1 and degradation by Cyp24A1 in the same cells. In healthy individuals, serum levels are normally 25–40 ng/mL (62–99 nM) for 25(OH)VD{SUB()}3{SUB}, and 20–45 pg/mL (48–108 pM) for 1,25(OH){SUB()}2{SUB}D{SUB()}3{SUB}.
In addition to the liver–kidney loop for the synthesis of 1,25(OH){SUB()}2{SUB}D{SUB()}3{SUB}, the immune system can independently generate bioactive VD through a distinct regulatory mechanism. It was initially recognized that activated macrophages in sarcoidosis, a form of calcified lung fibrosis, could generate abundant calcitriol.[3] Later it was found that normal macrophages under lipopolysaccharide (LPS) and interferon-gamma stimulation could also produce calcitriol.[4, 5] However, different from its synthesis in the kidney, the expression of Cyp27B1 in the monocytes and macrophages is not inhibited via feedback by serum calcium levels.[6] Although not well defined, it is generally believed that these two pools of 1,25(OH){SUB()}2{SUB}D{SUB()}3{SUB} may have distinct purposes. The canonical functions of VD, generated systemically through the liver and kidney loop, may facilitate intestinal absorption of calcium by mediating active calcium transport (calbindin) across the intestinal mucosa, which maintains calcium homeostasis in blood and allows for bone calcium deposition. On the other hand, calcitriol produced locally by immune cells may contribute to immune regulation, a protective measure for infection and immune regulatory functions. The 1-hydroxylase enzyme Cyp27B1 in the kidneys is induced by PTH in response to low calcium in the blood, while the isoenzyme outside the kidney is independent of PTH induction. That the same cyp27b1 gene with identical cis-regulatory elements is regulated differently in a tissue-specific manner is likely regulated by epigenetic determination (Fig. 1).
Figure 1. Two systems and pools of bioactive vitamin D, and their functions.
Genomic and non-genomic actions by VD signals
Calcitriol assumes its cellular functions through binding to the VD receptor (VDR), a member of the nuclear receptor family of transcription factors.[7] VDR has four distinct domains: a ligand-binding domain for calcitrol, a retinoid X receptor (RXR) binding domain, a DNA binding domain that recognizes VD response cis-elements, and an activation domain to bind other transcriptional cofactors. Upon ligand engagement, VDR undergoes phosphorylation, which allows for heterodimer formation with RXR. The ligand-bound heterodimer subsequently moves into the nucleus and binds to specific VDR-responding cis-elements, termed VDREs. The VDRE consensus sequence has been characterized as an A/GGG/TTCA motif, although there is considerable sequence diversity, and most genes regulated by calcitriol have multiple VDREs in their promoters. Upon binding to VDREs, the activated complex recruits co-activators or co-repressors that either promote or repress transcription of specific genes, respectively. At cellular levels, VDR signaling was found to inhibit cell cycle transition and promote differentiation.[8, 9]
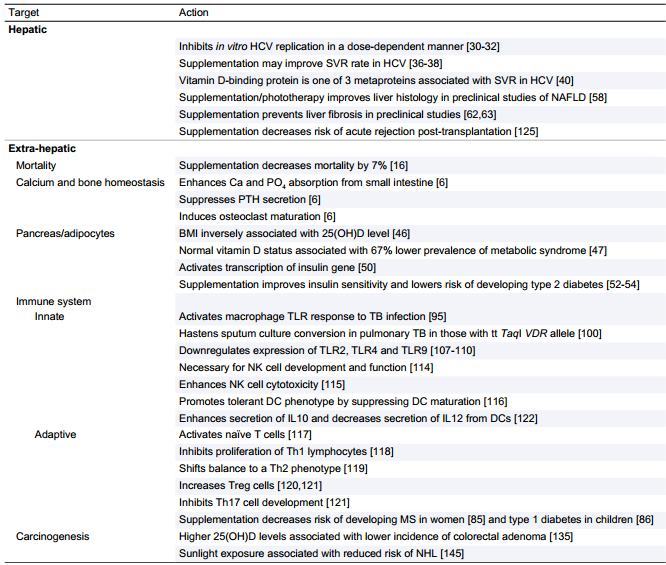
Genomic functions of calcitriol are best described by induction or suppression of its targeting genes (Table 1). Indeed, VD-regulated genes can be categorized into five groups. The first group including Cyp27A1, Cyp27B1, and PTH—which are suppressed by calcitriol—is related to VD synthesis, in addition to Cyp24A1, which is induced by calcitriol and responsible for its breakdown.[9, 10] The second group, including calbindin (a calcium-binding protein) and TRPV6 (a calcium channel), which are up-regulated by calcitriol, is related to calcium homeostasis.[11, 12] The third group, including cathelicidin and defensin beta, which are induced by VD, is related to immune defense.[13] The fourth group—including interleukin-2 (IL-2), IL-12, and IFN-gamma that are suppressed by calcitriol—is related to immune regulation and suppression.[14-16] Finally, the fifth group, which is related to bile acid metabolism including FGF15 (fibroblast growth factor 15 for murine), which suppresses Cyp7A1, a key enzyme for bile acid synthesis, and ASBT (apical sodium dependent bile acid transporter expressed in intestine for re-adsorption of bile acid), is up-regulated by calcitriol.[17, 18]
| Table 1. The targeting genes regulated by vitamin D, VDR targeting genes/sites |
Induced or suppressed | Functions of targeting genes |
| Parathyroid hormone (PTH), in parathyroid glands | Suppressed | Induces Cyp27B1 and suppresses 24-hydroxylation to enhance calcitriol synthesis under low calcium concentration in the blood |
| Cyp24A1 (VD-24-hydroxylase), in the kidney | Induced | Inactivates calcitriol to 1,24,25(OH){SUB()}3{SUB}D{SUB()}3{SUB}, which can be oxidized to calcitrol acid for secretion. |
| FGF15 (FGF19 for humans), by ileum | Induced | Suppresses bile acid synthesis in hepatocytes through induction of SHP, a transcriptional suppressor. |
| Cyp27B1 (VD-1alpha-hydroxylase) in the kidney | Suppressed | Restrains VD synthesis by feedback inhibition |
| Cyp3A4, in the liver | Induced | Catabolizes bile acid and xenobiotics, |
| MRP3 (multidrug resistance-associated protein 3), by the liver | Induced | Transports a wide range of substrates including anti-cancer drugs and bile acid |
| SULT2A1 (sulfo-conjugating phase II enzyme; sulfotransferase), in the liver | Induced | Detoxifies steroids, xenobiotics, and bile acid for secretion. |
| ASBT (apical sodium/bile acid co-transporter), in the ileum | Induced | Enhances bile acid re-adsorption from ileum, which may restrain bile acid synthesis in the liver via feedback inhibition. |
| Calbindin D9K (calcium binding protein/transporter), in the intestine and kidney | Induced | Transport of calcium across the enterocytes from the apical side |
| TRPV6 (calcium channel), in the intestine. | Induced | For the first step in calcium absorption in the intestine |
| Cathelicidin (LL37; hCAP - human), in the intestine and immune cells. | Induced | Antimicrobial peptides in macrophages and neutrophils in innate immune defense against invasive bacterial infection |
| Defensin beta 2 (cysteine-rich cationic low molecular weight antimicrobial peptide), by lung, skin, and innate immune cells. | Induced | Exhibits potent antimicrobial activity against Gram-negative bacteria and Candida, but not Gram-positive S. aureus |
| FasL/CD95L (ligand for Fas), in immune cells. | Suppressed | Reduces apoptosis |
| Interferon-gamma, in T cells. | Induced | Restrains immunity |
| Interleukin-12 (IL12), in the innate cells | Suppressed | Restrains Th1 activation by limiting IL-12, an essential cytokine from monocytes. |
| E-cadherin, enhanced in skin and epithelial cells. | Suppressed | Restrains epithelial to mesenchymal transition |
In addition to regulating gene expression/suppression, VD may also exert non-genomic actions, featured by rapid non-transcriptional regulation.[19] One study found that 1,25-dihydroxyvitamin D{SUB()}3{SUB} can induce a rapid and transient release of inositol triphosphate (IP{SUB()}3{SUB}) and diacylglycerol (DAG), which rapidly activate PKC to regulate Ca2+ fluxes through voltage-operated channels in myoblasts.[20] Importantly, the signaling transduction and rapid calcium flux function are independent of genomic transcription.[21] Indeed, a role for VD in calcium signaling in the muscle is supported by an early study showing that VD deficiency affects muscle relaxation and contraction.[22] While VDR is expressed at minimal levels in the mouse liver, it is abundant in the human liver.[23] Thus, any interpretation of the functions of VD in the liver from animal models should be taken cautiously. On the other hand, VDR is highly expressed in gastrointestinal epithelial cells of both mice and humans. Thus, one study showed that VD could induce FGF15 from the ileum, which may consequently target the liver to suppress Cyp7A1, a key enzyme for bile acid synthesis.[24]
VD in immunity
1,25-dihydroxlvitamin D is synthesized by innate and adaptive immune cells
The involvement of VD in immunology was first described almost 30 years ago, and since then, such functions have greatly been discovered for innate, adaptive, and regulatory immunity. Monocytes/macrophages isolated from patients with granulomatous disease, a type of lung fibrosis, can constitutively generate 1,25-dihydroxyvitamin D.[25] Similarly, monocytes isolated from normal human peripheral blood readily synthesize 1,25-dihydroxyvitamin D when treated with cytokines such as IFN-gamma or LPS.[4] These studies suggest that the synthesis of bioactive VD is an acute response to infection, which may in turn to restrain excessive response through immune regulation. Indeed, active VD inhibits CD40L-induced activation of human monocytes and expression of TNF-alpha and IL-1.[26] Moreover, in an experimental inflammatory bowel disease model, mice with disrupted VDR expression exhibited high colonic expression of TNF-alpha, IL-1, IL-12, and IFN-gamma, in addition to being extremely sensitive to innate injury, thus indicating the immune suppressive functions of VD.[27]
Antigen presenting cells express VDR and its signaling machinery
1,25-dihydroxyvitamin drives differentiation of monocytes into macrophages, indicating the expression of VDR and the necessary machinery for signal transduction.[28] Likewise, dendritic cells also express VDR.[29] Further studies showed that allogeneic T cells stimulated with 1,25-dihydroxyvitamin-primed dendritic cells were poor responders, suggesting that 1alpha,25-(OH)2D3 may modulate the immune system through inhibition of dendritic cell differentiation and maturation into potent antigen-presenting cells (APC).[30]
Antimicrobial peptides are induced by VD in innate immune cells
In addition to inducing CD14 for innate immunity, 1,25-dihydroxyvitamin D can also up-regulate antimicrobial peptides such as cathelicidin and beta-defensin by macrophages in the gastrointestinal track.[13, 31] In fact, activation of toll-like receptors in human macrophages can induce cathelicidin antimicrobial peptide, which is inhibited by either a VDR antagonist or a non-specific inhibitor against 1-hydroxylase activity, suggesting that de novo synthesis of 1,25-dihydroxyvitamin D is required for cathelicidin induction.[32] Induction of beta-defensin expression by 1,25-dihydroxylvitamin D requires additional activation of NF-kappaB sites, which are adjacent to its VDRE via a mechanism involving IL-1, suggesting converge of inflammatory signals and immune regulation.[33] Thus, VD-induced antimicrobial peptides in the gastrointestinal track may represent a typical example of host defense modulation of the innate immune system.
Immune suppressive functions of VD are featured by direct suppression of Th1 response
Th1 cells secrete pro-inflammatory cytokines, including interferon-gamma (IFN-gamma) and IL-2, and activate B cells to produce immunoglobulin IgG2a. A large body of evidence has demonstrated that VD/VDR can suppress Th1 lymphocytes by mechanisms such as down-regulation of IL-2, a key cytokine for T cell proliferation and activation.[34] Moreover, 1,25-dihydroxyvitamin D also suppresses IL-12 and IFN-gamma, the two major Th1 cytokines for acute immune responses.[35] In contrast, Th2 cells secrete IL-4 and IL-10, and induce the production of IgG1 and immunoglobulin E (IgE) by B cells, in a manner skewed to immune regulation. Interestingly, activated monocytes and dendritic cells from the innate immune system, and B cells from the adaptive immune system produce IL-12, which induces Th0 to become Th1 cells. Consequently, activated Th1 cells induce inflammation by generating IFN-gamma and TNF-alpha. For such regards, 1,25-dihydroxyvitamin D suppresses IL-12 production by monocytes and B cells, thus consequently restraining Th1 activation.[36]
VD up-regulates T-regulatory cells for immune regulation/tolerance
Epidemiologic studies have shown that low blood VD levels are associated with autoimmune diseases. In particular, the potential role of VD in immune regulation by induction of T-regulatory cells (Tregs) is under extensive scrutiny. For example, bioactive VD was found to potentiate Treg activity,[37] and low levels of 25-hydroxy VD in the circulation are associated with compromised Treg function and high incidence of multiple sclerosis.[38] Accordingly, one study showed that high levels of 1,25-dihydroxyvitamin D are relevant to Th2 skew and increased Tregs in multiple sclerosis.[39] In a skin immune lesion model, topical application of 1,25-dihydroxlvitamin D can suppress antigen-induced CD8+ cell activation through induction of antigen-specific Tregs.[40] Finally, to demonstrate direct induction of Tregs by VD, healthy volunteers were given high doses of VD supplement (VD3) and compared with a group receiving a placebo control. The authors found that the CD4 *CD25hiFoxP3 * Tregs cells were significantly increased, concomitant with increased endogenous synthesis of 1, 25-dihydroxyvitamin D{SUB()}3{SUB}.[41]
Direct regulation of Th2 cells by VD
Several lines of research have demonstrated that VDR activation promotes Th cell polarization by inhibiting Th1 and by augmenting Th2 cell development, thus inhibiting IFN-gamma and up-regulating IL-4, IL-5, and IL-10 production.[42] The VD-induced effects were largely mediated via IL-4, as IL-4 neutralization almost completely abrogated the observed augmented Th2 cell development after D3 treatment. Furthermore, increased expression of the Th2-specific transcription factors GATA-3 and c-maf correlated with increased production of Th2 cytokines after VD treatment. In addition, allergic asthma is tightly associated with Th2 cells. VDR knockout mice, however, failed to develop experimentally induced allergic asthma, suggesting an important role for VD signaling in the generation of Th2-driven inflammation.[43] On the other hand, 1,25-dihydroxyvitamin D can suppress Th2 skewed immune responses via naive Tregs.[44] Indeed, administration of 1,25-dihydroxyvitamin D significantly suppressed ovalbumin (OVA)-induced allergy through reduction of serum OVA-specific IgE levels, airway eosinophilia, and Th2-related cytokines.[45]
VD deficiency in chronic liver diseases and clinical trials
VD deficiency is frequently found in chronic liver diseases.[46] Active VD can suppress hepatic stellate cell activation in vitro and hepatic toxin-induced cirrhosis in a rat model.[47] However, it is still ambiguous as to what level is regarded as VD insufficiency or deficiency apart from its classic definition for calcium adsorption/deposition. Neither the VD standard for health liver function is defined, nor the threshold for chronic liver diseases is known. A working standard has been generally adapted from the Endocrine Society, which defined that 32 ng/mL should be used as the threshold for 25(OH)D sufficiency in patients with various disease conditions.[48]
VD in non-alcoholic steatohepatitis
Non-alcoholic fatty liver disease (NAFLD) is characterized by hepatic steatosis in patients who do not exhibit alcohol abuse or other known liver diseases. Non-alcoholic steatohepatitis (NASH) is a progressive form of NAFLD characterized by both hepatic inflammation and lipid excessiveness. NAFLD affects about 20–30% of the adult population and 8% of adolescents in many countries.[49] NAFLD is tightly associated with obesity, metabolic syndrome, insulin resistance, and type-II diabetes mellitus, which are related to VD deficiency or insufficiency.[50] In particular, serum 25-hydroxyvitamin D levels have been found to be inversely related to body mass index and body fat content, hypertension, insulin resistance, and diabetes mellitus.[51, 52] Importantly, the results from a clinical trial on obese adolescents showed that body fat content is significantly associated with VD deficiency or insufficiency.[53]
The mechanisms behind excessive lipid accumulation in NAFLD are still intriguing. An elegant study using stable isotopes to trace fatty acids in NAFLD patients found that about 60% of hepatic triglycerides come from serum non-esterified fatty acids from the diet, while about 26% triglycerides derive from de novo synthesis in the liver, indicating that abnormal triglyceride intake may contribute mostly to the biogenesis of NAFLD.[54] Increased intake by efficient emulsification of lipids by bile acids is probably the first step leading to excessive hepatic lipid accumulation. Therefore, we speculated that bile acid availability in the intestine after meal might contribute to NAFLD and even to NASH. Bile acids are tightly controlled at many levels, starting from their synthesis and catabolism in the liver, storage in the gallbladder and secretion after meal, re-adsorption in the ileum, and finishing with their degradation in the liver. At the molecular level, bile acids are subject to negative feedback control through the farnesoid X receptor and interactions with RXR.[55] VD plays an important role in controlling bile acid synthesis, secretion, and catabolism. For instance, VD was found to inhibit bile acid synthesis by suppressing Cyp7A1 through induction of intestinal FGF15, which in turn induces hepatic small heterodimer partner (SHP), a transcriptional suppressor targeting the Cyp7A1 gene.[24] VD also induces intestinal bile acid transporters for re-adsorption, resulting in feedback inhibition of bile acid synthesis.[56] Moreover, VD can induce Cyp3A4, a key enzyme for bile acid catabolism,[57, 58] indicating a potential role in lipid adsorption. Hence, we speculate that VD deficiency may exacerbate NALDF and NASH in part through insufficient negative regulation of bile acid bioavailability.
VD in alcoholic liver disease (ALD)
ALD is a major cause of chronic liver diseases and can lead to fibrosis and cirrhosis. The latest surveillance report, published by the National Institute on Alcohol Abuse and Alcoholism, reported that liver cirrhosis was the 12th leading cause of death in the United States, with a total of 29 925 deaths in 2007, 48% of which were alcohol related. Although not well addressed, VD deficiency is an issue in ALD. For instance, one study showed that among the patients with alcoholic cirrhosis, 85% had serum VD levels below 50 nmol/L, and 55% had levels below 25 nmol/L.[59] Questions are still open as to whether alcohol impairs VD adsorption or impedes 25- or 1-hydroxylation for synthesis of endogenous active VD. For ALD, it is possible that VD may modulate the early immune response through Th2 and Treg regulation. Equally possible is that VD may regulate the genes for alcoholic metabolism. Deficiency of VD thus may lead abnormal alcoholic catabolism and excessive TG accumulation in the liver; the subjects should be addressed.
VD in viral hepatitis
A growing body of clinical evidence has demonstrated the prevalence and risks of VD deficiency in patients suffering from chronic hepatitis C infection. Conversely, VD supplementation has been proposed as an adjunct to current standard cares for treatment of hepatitis C.[60] A clinical study found that 25(OH)D serum levels were significantly lower in chronic hepatitis C (25 μg/L) than in the controls (43 μg/L).[61] Expression levels of CYP27A1 correlated with 25(OH)D levels, but they were inversely related to necroinflammation. Moreover, low VD is linked to severe fibrosis and impaired sustained virologic response (SVR) in IFN-based therapy. One clinical trial showed that adding VD to the standard IFN plus ribavirin treatment significantly increased SVR in patients with chronic hepatitis C virus (HCV) genotype 1.[62] The SVR was defined as undetectable HCV-RNA at 24 weeks post-treatment. The increased SVR attained by VD treatment was found to be even better for patients infected with HCV genotypes 2 and 3.[63] Another clinical study showed that the levels of VD and of its active form were significantly lower in advanced liver disease (hepatic cirrhosis and/or carcinoma) patients.[64] Conversely, low VD is associated with inflammation, as shown by elevated IL-17 and IL-23 levels in advanced patients.
Regarding the underlying molecular mechanisms, an in vitro study showed that vitamin D3 remarkably inhibits HCV production in Huh7.5 hepatoma cells.[65] These cells express VD hydroxlases and can eventually generate calcitriol. Notably, treatment with calcitriol resulted in HCV inhibition through induction of IFN-beta. Intriguingly, HCV infection increased calcitriol production by inhibiting CYP24A1, implying that viral-induced immune tolerance may favor viral chronicity. A recent study surprisingly found that 25-hydroxyvitamin D3, but not 1,25-dihydroxyvitamin D3, is capable of reducing HCV by inhibiting infectious virus assembly.[66]
VDBP gene polymorphisms may also determine the VD levels. By examining HCV patients treated with a combination therapy of pegylated interferon alpha (PEG–IFN) plus ribavirin, one study found that good responses to the treatment were related to both the VD levels greater than 20 ng/mL and the wild-type VDBP polymorphisms.[67] VDR polymorphisms have also been associated with liver diseases, such as primary biliary cirrhosis.[68, 69] A recent clinical study measured the effects of 25-OH VD plasma levels and VDR polymorphisms on fibrosis progression in HCV patients. Results showed that the bAt(CCA)-haplotype was significantly associated with fibrosis progression.[70]
Conclusion
VD deficiency or insufficiency is well recognized for the association with variety of chronic degenerative diseases, including chronic hepatitis, viral persistence, ALD, NASH, and poor responsiveness for antiviral treatment. Among its multiple functions, immune modulation/regulation by VD is essential for tissue homeostasis and health physiologic response in addition to its job in calcium adsorption. At tissue and cellular levels, through its nuclear receptor, which is plentiful in immune cells and intestinal track, VD suppresses innate and adaptive immunity through inhibition of Th1, promoting Th2 response, and enhancing Tregs. Clinical trials have yielded striking results, showing that VD supplement can significantly enhance the SVR for IFN-based anti-HVC treatment. Additional clinical trials are initiated to confirm the therapeutic efficacy, which can be found in http://www.clinicaltrials.gov. VD deficiency is also prevalent in NFLD, NASH, and ASH, suggesting potential roles of VD in restraining the development of fatty liver diseases. Our recent work in line with others demonstrates that VD may restrain bile acid bioavailability through a variety of mechanism from
inhibiting bile acid synthesis in the liver,
promoting bile salt retention in the gallbladder,
re-adsorption in the gut, to its
breakdown in the liver.
A major adverse effect that limits large dose and long-term application of calcitriol is its potential to generate hypercalcemia. To such regard, searching for VD directives that retain effects on immune modulation with less impact on hypercalcemia is an endeavor for research and development.
References are in PDF at the bottom of this page
See also VitaminDWiki
Overview Liver and vitamin D contains the following summary
{include}
20 Most-visited pages in Liver category
{LISTPAGES}