Health benefits of non-burning exposure to UV light
 See also VitaminDWiki
* Health benefit of Sunlight is more than Vitamin D in the blood - many studies
* Low UVB (thus low Vitamin D) is linked to many diseases – Grant Jan 2016
* UV, sunshine, and vitamin D (87 charts) - Holick March 2013 = Author of editorial
* The risks and benefits of sun exposure – Holick Oct 2016 = Author of editorial
* Go ahead, soak up some sun (but don’t burn) – Holick July 2015 = Author of editorial
* Vitamin D protects DNA against UV skin damage – 5 studies 2012-2013
* UVB produced 8X more vitamin D as solar UV – July 2012
* It appears that UVA decreases the vitamin D generated by the UVB
* Less sun means more disease -Grant, Holick, Cannell, et al Feb 2015 co-author was author of editorial
* Time in sun (in Spain) to get 4,000 IU of vitamin D: half hour in July , 2 hours in October – Sept 2016
* Overview UV and vitamin D
* Sun and UV
* Optimize vitamin D from the sun
* Overview Suntan, melanoma and vitamin D
No – 10 minutes per day of sun-UVB is NOT enough contains the following summary
{include}
See also VitaminDWiki
* Health benefit of Sunlight is more than Vitamin D in the blood - many studies
* Low UVB (thus low Vitamin D) is linked to many diseases – Grant Jan 2016
* UV, sunshine, and vitamin D (87 charts) - Holick March 2013 = Author of editorial
* The risks and benefits of sun exposure – Holick Oct 2016 = Author of editorial
* Go ahead, soak up some sun (but don’t burn) – Holick July 2015 = Author of editorial
* Vitamin D protects DNA against UV skin damage – 5 studies 2012-2013
* UVB produced 8X more vitamin D as solar UV – July 2012
* It appears that UVA decreases the vitamin D generated by the UVB
* Less sun means more disease -Grant, Holick, Cannell, et al Feb 2015 co-author was author of editorial
* Time in sun (in Spain) to get 4,000 IU of vitamin D: half hour in July , 2 hours in October – Sept 2016
* Overview UV and vitamin D
* Sun and UV
* Optimize vitamin D from the sun
* Overview Suntan, melanoma and vitamin D
No – 10 minutes per day of sun-UVB is NOT enough contains the following summary
{include}
📝 Download Editorial PDF from VitaminDWiki
📝 Download Study PDF from VitaminDWiki
Can you have your cake and eat it too? The sunlight D-lema – EDITORIAL
DOI: 10.111l/bjd.15127, British Journal of Dermatology
M.F. Holick, Section of Endocrinology, Nutrition and Diabetes, Department of Medicine, Boston University Medical Center, Boston, MA 02118, U.S.A. mfholick@bu.edu
In this issue of the BJD, Felton et al.1 report the results of their elegantly designed study to determine the impact on vitamin D status and skin DNA damage for healthy adults with skin types 2 and 5 of low-level simulated summer sunlight exposure as would be experienced at a latitude in the U.K.
The photosynthesis of vitamin D has been occurring on this planet for more than 500 million years ever since phytoplankton were producing vitamin D during sun exposure.2 Although vitamin D’s function in early life forms is not well understood, recent evidence has suggested that vitamin D3 stabilizes membranes that may be important in regulating immune function.3
Throughout evolution, many organisms, including vertebrates, have depended on sun exposure for their vitamin D requirement. Our hairy dark-skinned ancestors in Equatorial Africa had both their hair and skin pigment as mechanisms to prevent sun-induced skin damage. As our ancestors evolved, they lost their protective hair, and in turn increased melanin production as an effective natural sunscreen. However, the increased amount of melanin in the skin still resulted in a small amount of vitamin D3-producing-ultraviolet (UV) B radiation to reach the basal layer, promoting the production of vitamin D3 throughout the epidermis, which was essential for the maintenance of skeletal health throughout life. As our ancestors migrated north and south of the equator, the increase in the zenith angle of the sun reduced the amount of vitamin D3-UVB-producing photons reaching the skin to produce an adequate amount of vitamin D3. To compensate for vitamin D deficiency, mutations occurred resulting in a reduction in skin melanin content for those who migrated to the farthest northern and southern regions including the Neanderthals who likely had a Celtic skin tone.4
However, the loss of skin pigment now permitted UVB-sensitive macromolecules, including DNA, to absorb the solar UVB radiation that penetrated the epidermis. This absorption caused thymidine dimerization and other alterations in the DNA structure, increasing the risk for the development of nonmelanoma skin cancer.5 The Surgeon General’s report from the United States and many dermatology societies have promoted abstinence from any direct sun exposure, which is thought to be a major contributor for the worldwide vitamin D deficiency epidemic.6
In support of this message was a recent study that reported on Danish adults exposed to high-intensity sunlight during a vacation in the Canary Islands. Peterson et al.7 not only observed improvement in their vitamin D status but also a significant and concerning cutaneous DNA damage as measured by increased urinary cyclobutane pyrimidine dimers (CPD), a surrogate for DNA damage. Thus, it was suggested that you could not have your cake and eat it too, i.e. take advantage of the beneficial effect of sun exposure for producing the vital vitamin D3 without significant DNA damage in the skin. However, from an evolutionary perspective, this makes little sense for survival of the species, i.e. the need to be dependent on solar UVB for bone development and health while at the same time increasing the risk for skin cancer. Female infants with infantile rickets would have had a flat pelvis with a small pelvic outlet making it difficult for normal child birthing. This is thought to be the driver for loss of skin pigmentation as people migrated north and south of the equator.8 Those who migrated into the far northern reaches of the European continent ultimately lost most of their skin pigment in order to permit the vitally important vitamin D-producing UVB radiation to enter the epidermis to produce vitamin D3.8
However, the skin of the Danes with skin types 1 and 2 was not designed to be exposed to high-intensity sunlight for an average of 38 h over 6 days in an environment that was much farther south from where their ancestors evolved. Felton et al.1 explored the possibility that those with skin type 2, whose skin had evolved to produce adequate vitamin D3 when exposed to sunlight at latitudes in the U.K., had also developed mechanisms to repair the damage caused by the same exposure.
They exposed healthy adults with little skin pigmentation (i.e. skin type 2) to low-level simulated U.K. June midday sunlight (equivalent to 13-17 min, six times weekly) and evaluated its effect on raising blood levels of 25-hydroxyvitamin D [25(OH)D; a measure of vitamin D status] and at the same time monitored various outcome measures related to cutaneous DNA damage. As expected, they observed a significant 49% increase in the circulating levels of 25(OH)D as a result of 7-dehydrocholesterol in the epidermis absorbing UVB radiation (290-315 nm) resulting in its conversion to previtamin D3. Previtamin D3 is then converted within a few hours by a membrane-enhanced isomerization to vitamin D3. Once formed, vitamin D3 travels to the liver and is converted to 25(OH)D3.8 However, as UVB is penetrating into the epidermis to form vitamin D3, it is also absorbed by DNA resulting in the formation of CPD and other pyrimidine photoproducts that if unrepaired have been associated with increased risk for nonmelanoma skin cancer.9 It also resulted in CPD-positive nuclei in keratinocytes for the exposed skin compared with photoprotected skin of the same volunteer demonstrating the UVB-induced consequence of the DNA absorbing this radiation.
However, 24 h after 6 weeks of exposure they observed significant clearing of the CPD-positive nuclei. This corresponded to undetectable levels of CPD in the urine and no change or accumulation in another marker for DNA damage from baseline, i.e. urinary 8-oxo-20-deoxyguasine (8-oxo-dG), a measure of oxidatively damaged DNA.
Felton et al.1 also conducted the study on Asians with skin type 5. They compared skin type 2 with type 5, and found that there was more DNA damage done to those with type 2, supporting that our ancestors who migrated further from the equator were at a disadvantage when it comes to UVB skin protection. As has been previously reported, increased skin- protecting pigmentation efficiently absorbs UVB radiation and therefore also reduces the number of photons absorbed by 7-dehydrocholesterol, resulting in a decrease in the effectiveness of the sun in producing vitamin D3,8 which they also observed by demonstrating a statistically insignificant increase in serum 25(OH)D levels in their Asian subjects.
The skin has a large capacity to produce vitamin D3. It has been estimated that exposure in a bathing suit to 1 minimal erythemal dose is equivalent to ingesting approximately 15 000-20 000 IUs of vitamin D.8 Furthermore, Maasai herders who have skin type 6 have circulating levels of 25(OH)D on average of 48 ng mL-1.10 Achieving these levels requires the ingestion of 3000-5000 IUs daily.8 It is likely that our hunter-gatherer equatorial ancestors exposed to daily sunlight were making this amount of vitamin D3 and at the same time their high melanin skin content prevented significant DNA damage. However, as our ancestors migrated to higher latitudes, the effectiveness of the sun for producing vitamin D was markedly diminished. A major target was the melanocortin 1 receptor (MRC1R), which regulates pigmentation in humans and other vertebrates. Impairment of its activity is associated with the pale Celtic skin types 1 and 2.3,4
To compensate for the lack of the natural melanin sunscreen, other mechanisms were developed to repair UVB-induced damage to the DNA in the skin to prevent skin cancer.9 Several enzymes (exinucleases) have been identified that specifically recognize UVB-induced DNA damage and excise the damaged portion with follow-up DNA ligase repair.11 The rapid disappearance of CPD-positive skin cells 24 h after the final UV exposure for both skin types 2 and 5 and the inability to detect urinary CPD after single and multiple exposures to simulated U.K. sunlight in the Felton et al.1 study provided strong evidence that these mechanisms are in place to reduce the risk of UVB-induced skin cancer. The reason for not observing any significant DNA damage in the Felton et al.1 study compared with the Danish study7 is due to the fact that the Danes were exposed to an excessive amount of sunlight that their skin was not adapted for.
Therefore, you can have your cake and eat it too. The World Health Organization recognizes that limited sun exposure is an important source of vitamin D3 and at the same time warns against sunburning. The results of the study by Felton et al.1 support the concept that sensible sun exposure that does not cause sunburning, and is appropriate for the person’s skin type, can prevent vitamin D deficiency and its negative health consequences with little concern about this exposure increasing risk for skin cancer. However, sun exposure beyond that recommended for each person’s skin type should be avoided either through covering the exposed area or using sunscreen to prevent the DNA damage that is associated with longer exposure as was evident in the Danish study.7
It is now recognized that there are several other photochemical processes that occur in the skin during exposure to sunlight.6 These include the production of nitric oxide and carbon monoxide, which can help lower blood pressure and reduce risk for heart disease, and beta endorphin, which improves the feeling of well-being reducing the risk for depression. In addition, sunlight stimulates keratinocyte proopiomelanocortin gene-enhancing adrenocorticotropin hormone (ACTH) production as well as enhancing gene expression of several cytokines, all of which help to modulate immune function to reduce risk for autoimmune diseases and cutaneous infections.6 The results from Felton et al.1 should provide healthcare regulators, especially those who have advocated sun abstinence because of their concern for increased risk for skin cancer, with a new perspective for how the sun and our skin work in concert to take advantage of the many beneficial effects of sensible sun exposure while minimizing risk for skin cancer.
References in PDF
Concurrent beneficial (vitamin D production) and hazardous (cutaneous DNA damage) impact of repeated low-level summer sunlight exposures*
British Journal of Dermatology (2016) 175, pp1320-1328
S.J. Felton,1 M.S. Cooke,2 R. Kift,3 J.L. Berry,4 A.R. Webb,3 P.M.W. Lam,5 F.R. de Gruijl,6 A. Vail7 and L.E. Rhodes1 lesley.e.rhodes@manchester.ac.uk
'Dermatology Research Centre, Institute of Inflammation and Repair, Faculty of Medical and Human Sciences, University of Manchester, Manchester Academic Health Science Centre, Salford Royal NHS Foundation Trust, Manchester, U.K.
2Oxidative Stress Group, Department of Environmental and Occupational Health, Florida International University, Miami, FL, U.S.A.
3School of Earth Atmospheric and Environmental Sciences, University of Manchester, Manchester, U.K.
4Department of Clinical Biochemistry, Manchester Royal Infirmary, Central Manchester NHS Foundation Trust, Manchester Academic Health Science Centre, Oxford Road, Manchester, U.K.
5Oxidative Stress Group, Department of Cancer Studies and Molecular Medicine, University of Leicester, Leicester, U.K.
6Department of Dermatology, Leiden University Medical Centre, Leiden, the Netherlands
7Centre for Biostatistics, Institute of Population Health, University of Manchester, Manchester Academic Health Science Centre, Salford Royal NHS Foundation Trust, Manchester, U.K.
Summary
Background The concurrent impact of repeated low-level summer sunlight exposures on vitamin D production and cutaneous DNA damage, potentially leading to mutagenesis and skin cancer, is unknown.
Funding sources: This project was in part funded by Cancer Research U.K., project C20668/A10007. The funder had no involvement in the study design, data collection, data analysis, manuscript prepara tion or publication decisions.
- Plain language summary available online DOI 10.111l/bjd.14863
Objectives This is an experimental study (i) to determine the dual impact of repeated low-level sunlight exposures on vitamin D status and DNA damage/repair (via both skin and urinary biomarkers) in light-skinned adults; and (ii) to compare outcomes following the same exposures in brown-skinned adults.
Methods Ten white (phototype II) and six South Asian volunteers (phototype V), aged 23—59 years, received 6 weeks’ simulated summer sunlight exposures (95% ultraviolet A/5% ultraviolet B, 1-3 standard erythemal doses three times weekly) wearing summer clothing exposing ~35% body surface area. Assessments made were circulating 25-hydroxyvitamin D [25(OH)D], immunohisto- chemistry for cyclobutane pyrimidine dimer (CPD)-positive nuclei and urinary biomarkers of direct and oxidative (8-oxo-deoxyguanosine) DNA damage.
Results Serum 25(OH)D rose from mean 36-5 ± 13-0 to 54-3 ± 10-5 nmol L-1 (14-6 ± 5-2 to 21-7 ± 4-2 ng mL-1) in phototype II vs. 17-2 ± 6-3 to 25-5 ± 9-5 nmol L-1 (6-9 ± 2-5 to 10-2 ± 3-8 ng mL-1) in phototype V (P < 0-05). Phototype II skin showed CPD-positive nuclei immediately postcourse, mean 44% (range 27—84) cleared after 24 h, contrasting with minimal DNA damage and full clearance in phototype V (P < 0-001). The findings did not differ from those following single ultraviolet radiation (UVR) exposure. Urinary CPDs remained below the detection threshold in both groups; 8-oxo-deoxyguanosine was higher in phototype II than V (P = 0-002), but was unaffected by UVR.
Conclusions Low-dose summer sunlight exposures confer vitamin D sufficiency in light-skinned people concurrently with low-level, nonaccumulating DNA damage. The same exposures produce minimal DNA damage but less vitamin D in brown-skinned people. This informs tailoring of sun-exposure policies.
.
What's already known about this topic?
Repeated low-level exposures to simulated U.K. sunlight can produce vitamin D sufficiency in light-skinned people, but the concurrent impact on cutaneous DNA damage is unknown.
What does this study add?
- Low-level simulated sunlight exposures in people of skin phototype II conferred vitamin D sufficiency concurrently with DNA damage, which showed partial clearance at 24 h and no evidence of accumulated damage after 6 weeks of exposures.
- The same exposures produced minimal DNA damage but less vitamin D in brownskinned people (phototype V).
- The findings are informative for sun-exposure guidance.
Solar ultraviolet radiation (UVR) exposure has the established benefit to health of vitamin D synthesis, while skin cancer is a major hazard. Studies using various protocols have examined the impact of single- and repeated-dose UVR on vitamin D status,1 5 but research examining accompanying UVR-induced DNA damage is scarce. Recently, the impact of high-intensity UVR exposures attained through a sunbathing holiday (Canary Islands, 28°N) on circulating 25-hydroxyvitamin D [25(OH) D] and cyclobutane pyrimidine dimer (CPD) excretion in urine, as a proxy for UVR-induced cutaneous DNA damage, was explored in white individuals.5 There were increases in both vitamin D status and urinary CPD, and the conclusion was made that under high-level UVR exposure conditions, the vitamin D benefit is inevitably derived at the cost of DNA damage. However, this might differ with UVR exposure pattern and dose, and between phototypes.6,7
Skin cancer is prevalent and causes a substantial health burden in white populations. The main exogenous risk factor, UVR, is a carcinogen, initiating DNA damage and suppressing skin immunity.8 UVB induces pyrimidine (6-4) pyrimidone photoproducts9 and CPDs,10 the dominant mutagenic form of direct UVR-induced DNA damage,11 with thymine-containing dimers being most common.10 If not repaired, these photoproducts form the ‘UVB signature’ mutations present in skin cancers.12 Recently, UVA was also shown to induce thymine- containing dimers in human epidermis in vivo.10,13 UVR also induces oxidatively generated damage to nucleic acids.14 UVR- induced DNA damage stimulates melanogenesis, although this provides only modest protection against further UVR damage.15,16 Urinary excretion of UVR-induced DNA damage products may act as a convenient proxy for cutaneous DNA damage;17 however, to date, skin and urinary damage have not been directly compared.
UVB triggers conversion of 7-dehydrocholesterol to previtamin D, the body’s principal vitamin D source, with usually only small amounts obtained from diet.18 Vitamin D undergoes hepatic hydroxylation to 25(OH)D, the major circulating form and the current best indicator of vitamin D status, and subsequent renal hydroxylation to active 1,25-dihydroxyvitamin D. There is associative evidence of diverse health benefits of vitamin D,19 21 while its established benefit is musculoskeletal, including prevention of rickets and osteomalacia.22,23 Public health guidance recommends sun protection in individuals at high risk of skin cancer,24 while also considering vitamin D benefit. It is generally assumed that regular brief sun exposures to skin produce adequate vitamin D.25 Guidance is geared for light-skinned individuals, and is supported by an intervention study in 109 white patients where simulated low-level sunlight exposures, while they were casually dressed, produced vitamin D sufficiency, defined as 25 (OH)D > 50 nmol L-1 (20 ng mL-1).1
The objectives of this study were to examine the impact on cutaneous DNA damage/repair (skin and urinary biomarker assessment) alongside 25(OH)D gain with regular low-level UVR exposures, in both white- and brown-skinned people. We exposed 10 white and six South Asian volunteers to a simulated summer’s brief exposures (95% UVA/5% UVB, three times weekly for 6 weeks). Skin biopsies were examined for CPD-positive nuclei, for induction by a single 1 ■ 3 standard erythemal dose (SED) exposure, accumulation over 6 weeks’ UVR exposures, and clearance 24 h postcourse. Urine was analysed for CPD and 8-oxo-deoxyguanosine (8-oxo-dG) DNA damage.26 Through performance under known exposure conditions, the data gained are informative for sun-exposure guidance.
Patients and methods
Patients
This was an experimental study in healthy volunteers. People of phototype II (white skin, sunburns easily, tans minimally) and phototype V (South Asian, brown skin), aged i860 years, from Greater Manchester, U.K. were recruited by
advertisement (January 2010). Exclusion criteria were a history of skin cancer/photosensitivity, use of sunbed/sunbathing within 3 months, taking photoactive medication/ vitamin D supplements, and pregnancy or breastfeeding. The North Manchester Research Ethics Committee provided ethical approval (reference 09/H1014/73). The study adhered to the Declaration of Helsinki; the volunteers gave written, informed consent.
Minimal erythemal dose assessment
Individuals’ minimal erythemal doses (MEDs) were assessed precourse, as the lowest UVR dose producing a visually discernible erythema 24 h post-UVR. A geometric series of 10 doses (7-80 mJ cm-2 for phototype II; 26-6-271 mJ cm-2 for phototype V) of erythemally weighted UVR was applied to buttock skin using a Waldmann UV236B unit with CF-L 36W/UV6 lamps (peak emission 313 nm, range 290-400 nm; Waldmann GmbH, Villingen Schwenningen, Germany).
Simulated summer sunlight exposures
Volunteers were given a 6-week course of UVR exposures, concordant with the length of the U.K. school summer holiday, when the population is most exposed to sunlight, as described previously.1 A Philips HB588 Sunstudio irradiation cabinet (Philips, Eindhoven, the Netherlands) delivered whole-body UVR exposure after fitting with alternating Arimed B (Cosmedico GmbH, Stuttgart, Germany) and Cleo Natural (Philips) fluorescent tubes, providing UVR emission close to U.K. summer sunlight (95% UVA: 320-400 nm, 5% UVB: 290-320 nm). Cabinet emission was characterized using a DTM300 spectroradiometer (Bentham, Reading, U.K.) and monitored using an Ocean Optics S2000 spectroradiometer (Ocean Optics, Dunedin, FL, U.S.A.). Wearing protective eye goggles, standardized T-shirts and knee-length shorts, volunteers lay prone, exposing ~35% skin surface in total.
UVR exposures of 1-3 SED were given three times weekly in January and February, when ambient UVB is negligible at U.K. latitudes and people are at trough vitamin D status,27 with exposure time adjusted to maintain constant dosing.28 Doses took ~6-5 min to administer, equating to 13-17-min exposure to U.K. June midday sunlight exposure six times weekly, which takes account that (i) when horizontal, ventral and dorsal surfaces are exposed sequentially in sunlight, not simultaneously as in the cabinet; and (ii) in daily life, postures range from horizontal to vertical randomly orientated to the sun.29 To compare UVR-exposed/protected sites, a 10 x 10cm2 aperture was made in the shorts material over one buttock; the contralateral buttock was covered with UVR-opaque material.
Dietary vitamin D logs
Volunteers completed daily dietary logs of vitamin D-fortified foods, and six key food categories - cheese; butter, margarine and oily spreads; milk and milk-containing products; red meat; oily fish; and eggs and egg dishes - during the first and last study weeks.30 Vitamin D content was obtained from food package labelling and McCance and Widdowson’s The Composition of Foods.31
Vitamin D, parathyroid hormone and serum biochemistry
Blood samples were taken weekly, and serum stored at -20 °C until study completion. Serum 25(OH)D was measured by high-performance liquid chromatography-UV, as reported previously.32 The laboratory was accredited to ISO 9001:2008 and 13485:2003 standards, and certified proficient by the national vitamin D quality assurance scheme (DEQAS). Parathyroid hormone was measured at the beginning and end of the course, and serum biochemistry was analysed.1 Deficiency and sufficiency cut-offs for 25-(OH)D levels were defined as 25 nmol L-1 (10 ng mL-1) and 50 nmol L-1 (20 ng mL-1), respectively.23,33,34
Skin colour measurements
UVR-exposed and protected buttock skin colour was measured at baseline and weekly (CM-2500d spectrophotometer; Konica Minolta, Tokyo, Japan). Triplicate standard Lab* data were recorded.35 The individual typology angle (ITA) was calculated as the vector direction in the Lb plane, as arctangent (L-50)/b x (180/p).36 38
Cutaneous sampling
Following the UVR course, all participants had four 4-mm punch biopsies taken from buttock skin under the following conditions: photoprotected skin, immediately after 1 x 1-3 SED, immediately after 18 x 1-3 SED, and 24 h following the 18 exposures. Biopsies were formalin fixed and paraffin embedded for histological analysis.
Cutaneous cyclobutane pyrimidine dimer immunostaining
Immunostaining was performed using a modification of the method of Tewari et al.13 4-pm sections were treated with 0-1% trypsin; hydrogen peroxide (0-3% in methanol) was added to inhibit endogenous peroxidase; and blocking buffer (Vector Laboratories, Peterborough, U.K.) was added, followed by monoclonal antibody incubation (TDM-2, 1 : 2000; CosmoBio, Tokyo, Japan).39 The primary antibody was omitted from one slide/staining cycle as a negative control. Slides were incubated with biotinylated secondary antibody before addition of ABC solution, developed with Vector SG solution and counterstained with Nuclear Fast Red (Vector Laboratories) before dehydration and mounting. Images were scanned (Panoramic 250 Flash II; 3DHISTECH Ltd, Budapest, Hungary) and analysed for epidermal thickness and area (Image J 1.48; National Institutes of Health, Bethesda, MD, U.S.A.). Positively staining nuclei were counted per high-power field (HPF) (original magnification x40; 3 HPFs per section, 9 HPFs per slide). The researcher (S.J.F.) was blinded to the slide identity.
Urinary analyses for DNA damage
First-void urine samples were collected daily (Monday to Friday) during week 1 to assess for early impact, and then weekly to assess for accumulation of DNA damage. These were stored at —20 °C until processing.
Quantification of urinary 8-oxo-deoxyguanosine
Samples were analysed for 8-oxo-dG using ultrahigh-performance liquid chromatography (UHPLC)-tandem mass spectroscopy as described previously.40 The results were normalized using urinary creatinine.
Quantification of urinary thymine dimers
We developed a UHPLC-MS/MS assay for cis, syn T<>pT in urine, which benefits from stable isotope-labelled internal standardization, is more rapid than the HPLC32P-postlabelling method, avoids the need for 32P, and provides absolute quantification, unlike enzyme-linked immunosorbent assay. CPDs are removed from DNA by nucleotide excision repair, as a lesion-containing single-stranded oligomer approximately 2432 nucleotides long.41 These oligomers are subject to 5'?3' exonucleolytic attack, generating lesion-containing 6- and 7- mers, with some 2-mers. The current methodology for measuring CPDs in urine is HPLC prepurification followed by 32P postlabelling. This approach quantifies the dimer as a dinucleotide monophosphate (the dimerized form of thymidylyl- 3'-5'-thymidine, T<>pT).42 However, potential exists for dimers to be present in urine as other oligomeric forms. Therefore we adopted two approaches: the first quantifies T<>pT, and the second utilizes formic acid hydrolysis of urine to render all oligomeric forms down to the nucleobase form of the dimer (thymine-thymine dimer, T<>T). These methods are detailed in Appendix S1 (see Supporting Information).
Statistical analyses
Paired and unpaired t-tests, repeated-measures analyses and linear regressions were performed using SPSS statistical software (version 21.0.0; IBM, Armonk, NY, U.S.A.) and Graph- Pad Prism (version 6; GraphPad Software Inc., La Jolla, CA, U.S.A.). Ratio measures, logarithmically transformed to make them normally distributed, were considered statistically significant at P < 0-05.
Results
Volunteers
The volunteers were compliant with the study procedures and all completed the study. Table 1 displays their baseline
Table 1 Patient demographics (in PDF
Values are the mean ± SD unless stated otherwise. MED, minimal erythemal dose; 25(OH)D, 25-hydroxyvitamin D. aThe normal parathyroid hormone (PTH) range is 0-8-3-9 pmol L—1.
characteristics; general serum biochemistry was normal. Baseline serum parathyroid hormone appeared lower (nonsignificantly) for phototype II than phototype V, and did not change significantly. Dietary vitamin D was low, with 80% of phototype II and 83% of phototype V volunteers ingesting < 5 pg per day, and was constant between weeks.
Serum 25-hydroxyvitamin D gain
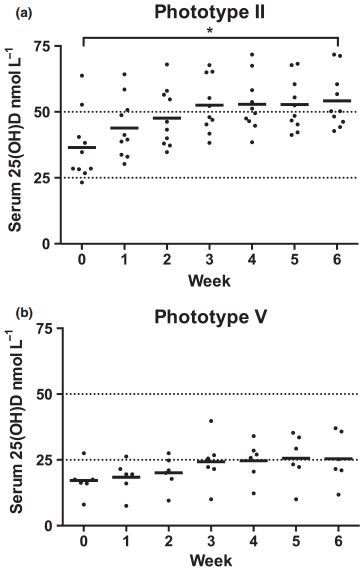
The 6-week course produced a greater mean serum 25(OH)D gain in phototype II volunteers: 17-8 ± 4-8 nmol L—1 vs. 8-3 ± 10-5 nmol L—1 for phototype V (P < 0-05; Fig. 1). The gain was inversely associated with baseline 25(OH)D for phototype II (R2 = 0-4; P = 0-049) but not phototype V. However, the proportional gain in 25(OH)D from baseline was almost identical, with a mean increase of 49% in phototype II, from 36-5 ± 13-0 at baseline to 54-3 ± 10-5 nmol L—1 at course end, and 48% from 17-2 ± 6-3 to 25-5 ± 9-5 nmol L—1 in phototype V. The post-UVR level was positively associated with baseline 25(OH)D (P < 0-001), consistently with previous studies.1,43
Skin darkening
At baseline, the mean L* (skin lightness) was 69 ± 2-8 in phototype II volunteers and 41 ± 12-8 in phototype V, with mean ITAs of 52 ± 5-7° and —22 ± 33-3°, respectively. The 6-week exposures produced significantly greater darkening in volunteers with phototype V than in those with phototype II, as indicated by the reduction in L* (P = 0-02), although this did not reach significance for ITA. ITA decrease (darkening) was positively associated with 25(OH)D gain for phototype II (R2 = 0-54, P = 0-016) but not phototype V volunteers, in whom there was wide interindividual variation in ITA and less 25(OH)D gain.
(a) Phototype II
Cutaneous cyclobutane pyrimidine dimers
Skin-section examination showed that UVR did not induce epidermal thickening in either phototype (data not shown). In the absence of UVR exposure, no CPDs were detectable in any individual (Fig. 2a, e). One 1-3-SED exposure caused a range of CPD levels in phototype II individuals (median count 200 CPD-positive nuclei mm-2, range 16-5—284; Fig. 2b), while only two phototype V volunteers showed any evidence of CPDs (counts of 4 and 16 CPD-positive nuclei mm-2; Fig. 2e). Skin receiving cumulative UVR (18 x 1-3 SED) showed elevated CPD-positive nucleus counts in phototype II (median 234 nuclei mm-2, range 125—314; Fig. 2c) vs. phototype V (median 12 nuclei mm-2, range 0—148, P < 0-001). No significant difference was seen in CPDs after cumulative vs.single exposure for either phototype. At 24 h after 6-week exposures, phototype II volunteers had cleared a mean 44% (range 27—84%) of their cutaneous CPD-positive nuclei, while those with phototype V had cleared 97% (range 84-100%, P < 0-001; Fig. 2d, e). Volunteers with phototype II showed a positive association of induction of CPD-positive nuclei with baseline ITA (R2 = 0-49; P = 0-02), but weak, nonsignificant associations with baseline L* (R2 = 0-29), age (R2 = 0-33) and 25(OH)D gain (R2 = 0-23).
Urinary DNA damage
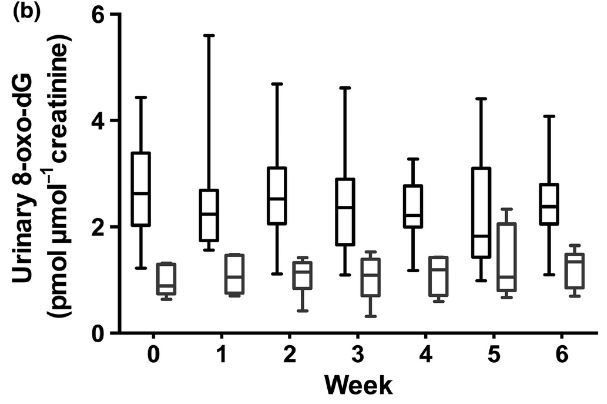
CPDs (T<>T and T<>pT) were undetectable for both phototypes, at baseline and after the UVR course. At baseline, phototype II volunteers had higher urinary 8-oxo-dG (mean 2-72 ± 0-97 pmol jimol-1 creatinine) than phototype V (mean 0-96 ± 0-28 pmol jimol-1 creatinine), P < 0-001, with no significant increase during any of the days measured in week 1 (Fig. 3a). Moreover, while 8-oxo-dG levels were higher in phototype II volunteers at all time points (repeated- measures analysis, P = 0-001; Fig. 3b), there was no accumulation in urinary 8-oxo-dG over the 6-week course.
Discussion
To our knowledge, this is the first study examining the benefits and cutaneous DNA damage/repair effects of vitamin D concurrently following low-level UVR exposures. Employing radiation similar to summer solar UVR emission and protocols simulating repeated casual exposures, UVR doses were equivalent to 13-17 min of U.K. June midday exposure, on most days of the week (latitude 53-5°N).29 Such exposures have been assumed to provide adequate vitamin D status, and were shown to provide serum 25(OH)D levels equating to sufficiency (50 nmol L-1)23 in white individuals.1 Assessment of concurrent DNA damage outcome (cutaneous and urinary) has awaited exploration.
We have now demonstrated that low-level exposures readily induced CPDs in keratinocytes in white skin (phototype II) and, to a much lesser extent, in South Asian skin (brown, phototype V) in vivo. Induction was significantly positively associated with skin pallor (baseline ITA), consistent with a recent ex vivo human skin study.38 Comparison of DNA damage induced by one exposure to 1 - 3 SED with that following 18 doses revealed close similarity. There was no evidence for regular low-level exposures leading to DNA damage accumulation, indicating effective repair between exposures.
Sheehan et al.44 described accumulation of CPD-positive nuclei with repeated 0-65-MED exposures in skin phototypes II and IV. However, this involved an MED-adjusted dose, not and absolute UVR dose, and exposures at shorter intervals (Monday to Friday for 2 weeks). Another human study reported that it took 48-72 h for CPD-positive nucleus levels to return to baseline following a single higher (1-2-MED) exposure.45 In human keratinocytes in vitro, following low- level UVB exposure (8 mJ cm-2, around twofold lower than
• Phototype II * Phototype V
the phototype I MED) on eight consecutive days, very few CPD-positive nuclei had been repaired 24 h post-UVR.46 Similarly, mice given repeated low-level UVB (0-5 kJ m-2 every 24 h for 40 consecutive days) showed that CPD repair lagged behind formation, leading to damage accumulation.9 The low- level UVR we employed may cause insufficient DNA damage to overwhelm repair,44,47 or the 48-h intervals between exposures could provide sufficient repair time. It is also feasible that repair mechanisms are upregulated by repeated low-level exposures.
As CPD persistence can lead to mutagenesis, and repair kinetics in human skin are most rapid within 24 h,48 we
Fig 2. Representative epidermal DNA damage in individuals with phototype II and V skin under varying conditions of ultraviolet radiation (UVR) exposure. Cyclobutane pyrimidine dimer (CPD)- positive nucleus staining (black arrow) from a volunteer of phototype II (left column) and phototype V (right column). Original magnification X40. (a) Photoprotected skin; (b) immediately
following one 1-3 standard erythemal dose (SED) exposure; (c) immediately following the completion of the 6-week simulated summer sunlight exposures; (d) 24 h after the completion of the 6- week simulated summer exposures. (e) CPD-positive nucleus counts in volunteers with skin phototype II (circles; n = 10) and V (triangles; n = 6). DNA damage was absent from photoprotected skin in both groups. The median CPD-positive nucleus counts were significantly higher in phototype II than V immediately following a single UVR exposure, after the 6-week course of cumulative UVR exposures, and 24 h following the cumulative exposures (P < 0-001 for all). In both phototypes, the 6-week simulated summer sunlight exposures caused no statistically significant difference in CPD-positive nuclei compared with a single 1-3-SED exposure. Horizontal bars denote the median. Viable epidermal thickness measurements did not differ between the two phototype groups, and were unchanged by the simulated summer sunlight exposures. *P < 0-001.
quantified CPD-positive nuclei in biopsies taken 24 h post- UVR. In phototype II skin, a mean 44% of CPD-positive nuclei were cleared vs. virtually all (97%) in phototype V, where the initial level of damage was much lower. The cumulative UVR study of Sheehan et al.44 also found more complete repair in skin type IV than II at 1 week post-UVR. The decrease in CPD-positive nuclei we observed at 24 h showed significant interindividual variation within phototype II skin (27-84%). From a human health perspective, it was encouraging that CPDs did not accumulate over the UVR course; nevertheless, a substantial proportion of damaged cells were still present 24 h post-UVR, and the potential remains for mutagenesis after each DNA-damaging event.
Interestingly, following both single and repeated (18 sessions) low-level UVR exposures, urinary CPDs remained below the detection limit, and oxidatively damaged DNA did not increase from baseline, in either phototype. Concurrent skin-section analysis confirmed CPD induction, but lack of urinary CPD detection suggests that the damage was relatively small, and/or the number of cells affected was insufficient to generate a signal in urine. This conclusion is supported by the urinary 8-oxo-dG findings. Our previous study showed that urinary 8-oxo-dG increases 4 days following single, whole- body suberythemal (15 J cm-2) UVA exposure in vivo,17 suggesting that our levels of UVR exposure (reflecting the UVR dose and surface area exposed) were insufficient to induce urinary 8 -oxo-dG, a sensitive biomarker of oxidative stress. Intriguingly, phototype II skin had greater urinary 8-oxo-dG than phototype V at all time points, implying a non-UVR explanation, such as differences in metabolism, repair and/or antioxidant intake; this warrants future exploration.
Fig 3. Urinary 8 -oxo-deoxyguanosine (8 -oxo-dG) damage (pmol pmol-1 creatinine) in volunteers with skin types II and V. Urinary 8 -oxo-dG concentrations (a) daily for the first 5 days of week 1 and (b) weekly during the 6-week simulated summer. Patients of phototype V (n = 6) had significantly lower 8 -oxo-dG levels than those of phototype II (n = 10) both in the first 5 days (including prior to exposure) and during the study (P = 0-001 and P = 0-002, respectively, repeated measures). However, no increase in urinary oxidative DNA damage was seen at any of the time points following a single UVR exposure during the first 5 days, and no accumulation occurred over the 6-week course. The data shown are the median, interquartile range and full range.
Studies examining the impact of melanin on vitamin D synthesis in vivo show conflicting results,2,4,6,7,49 potentially through differences in skin site, baseline 25(OH)D, UVR dose and UVR spectrum.50 We found that most phototype II participants reached vitamin D sufficiency [25(OH) D > 50 nmol L-1], which is consistent with our larger sample of white volunteers.1 Over half of the South Asian volunteers achieved sufficiency, attaining > 25 nmol L-1, but none reached > 50 nmol L-1, as in a previous investigation in 15 South Asian patients.30 In addition to the higher constitutive pigmentation in phototype V skin, they had significantly greater skin darkening during the UVR course than those with phototype II, and this may be responsible for the lower 25 (OH)D gain/plateau in phototype V. Facultative pigmentation includes involvement of higher epidermal levels,51 limiting UVB penetration to 7-dehydrocholesterol and hence initiation of vitamin D synthesis.
A positive association between urinary T < > pT and 25(OH) D gain was reported following intense UVR exposures (mean 60-101 kJ m-2) during sun/ski holidays in individuals with skin phototypes I-IV.5 Liljendahl et al.52 also identified significantly increased urinary T < > pT 3-5 days after 2 days’ beach sunbathing in Sweden, with urinary DNA damage strongly correlating with personal UVR dosage (up to 1400 J m-2). These high-dose exposures contrast with our brief suberythemal exposures, where the association between cutaneous CPD- positive nuclei and 25(OH)D gain was weak and nonsignificant. Building on the present study, application of a dose range of low-level UVR exposures could assess whether there are doses where vitamin D benefit is gained with minimal DNA damage in light-skinned adults. Similarly, a dose range of higher-level exposures4 could examine whether brownskinned individuals can achieve higher serum 25(OH)D gain, still with limited DNA damage.
The main strength of this study is the original, concurrent examination of cutaneous CPDs with urinary DNA damage biomarkers and 25(OH)D gain, following low-level UVR exposure. This simulation of northerly-latitude summer sunlight exposures employed UVR emission close to that of midday sunlight, and examined 25(OH)D gain after repeated exposures to commonly exposed skin sites. Completion of dietary logs indicated no alteration in vitamin D intake over the study. Future studies may explore the findings in a wider range of phototypes, using differing patterns of UVR and natural sunlight exposure.
Our findings indicate tailoring of public health policies on safe sun exposure for different phototypes. Brown-skinned individuals who experience almost negligible DNA damage but generate low amounts of 25(OH)D could be advised on less limited sun-exposure practice,4 while caution is required for phototype II individuals, as unrepaired cutaneous DNA damage was seen at 24 h following even the low UVR doses employed, in these easily burning individuals.
References in PDF 📄 Download the PDF from VitaminDWiki
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s website:
Appendix S1. Supplementary materials and methods.