T1DM, toll-like receptors and vitamin D in rats
The role of toll-like receptors and vitamin D in diabetes mellitus type 1 - a review.
Scandinavian Journal of Immunology ; Received Date : 24-Mar-2014 Revised Date : 28-Apr-2014 Accepted Date : 04-May-2014
Daria M. Adamczak1,3 daria.m.adamczak@gmail.com, Jan K. Nowak2, Magdalena Frydrychowicz3, Mariusz Kaczmarek3, Jan Sikora3
1 Poznan University of Medical Sciences, Clinical Hospital No. 1, Poznan, Poland
2 Poznan University of Medical Sciences, Department of Pediatric Gastroenterology and Metabolic Diseases, Poznan, Poland
3 Poznan University of Medical Sciences, Department of Clinical Immunology, Poznan, Poland * - equal contribution
It is widely accepted that type 1 diabetes mellitus (T1DM) is an autoimmune disease resulting from an interaction between immunologic, genetic, and environmental factors. However, the exact mechanism leading to the development of T1DM remain incomplete. There is a large body of evidence pointing towards the important role of toll-like receptor (TLR) activation and vitamin D deficiency in T1DM pathogenesis. In this article we review the available data on the influence of TLRs' level of activation and vitamin D status on the risk of the development of T1DM in humans and rodent models. We also summarized the current information regarding the interactions between TLRs' level of activation, vitamin D status, and various environmental factors, such as enteroviral infections, the gut microbiota, and breastfeeding substitution, among others. Our results stipulate that vitamin D seems to protect against T1DM by reducing the TLRs' level of activation.
Abbreviations
1,25(OH)2D - 1,25-dihydroxyvitamin D
25(OH)D - 25-hydroxyvitamin D
APC - Antigen presenting cell
B6/RIP-B7.1mouse - A mouse expressing RIP-B7.1 transgene
BBDP rat - Biobreeding diabetes-prone rat
BBDR rat - Biobreeding diabetes-resistant rat
CCL - C-C motif chemokine ligand
CD - Cluster of differentiation
CI - Confidence Interval
CL097 - A derivate of the imidazoquinoline compound R-848; TLR7/ TLR8 ligand
CMV - Cytomegalovirus
CpG - An unmethylated sequence of bacterial
DNA recognized by TLR9
CV-B4 - Coxsackie virus B4
CXCL - C-X-C motif chemokine ligand
DAMP - Damage-associated molecular pattern
DC - Dendritic cell
dsRNA - Double-stranded RNA
EBV - Ebstein-Barr virus
HMGB1 - High-mobility group protein B1
ICA - Islet cell antibodies IFN –
Interferon IL - Interleukin KRV –
Kilham rat virus LCMV -
Lymphocytic choriomeningitis virus
LCMV-GP - The envelope glycoproteins of LCMV
LPS - Lipopolysaccharide LTA - Lipoteichoic acid
MAP kinase - Mitogen-activated protein kinase
MHC - Major Histocompatibility Complex
mRNA - Messenger RNA
MyD88 - Myeloid differentiation primary response gene (88)
NF-kB - Nuclear factor kappa-light-chain-enhancer of activated B cells
NOD mice - Non-obese diabetic mice
OR - Odds ratio
OVA - Ovalbumin
PAMP - Pathogen-associated molecular pattern
PBMC - Peripheral blood mononucleated cell
Poly(I:C) - Polyinosinic:polycytidylic acid
PRR - Pattern recognition receptor
R-848 - An imidazoquinoline compound; TLR7/TLR8 ligand
SNPs - Single-nucleotide polymorphisms
ssRNA - Single-stranded RNA
STZ - Streptozocin
T1DM - Diabetes mellitus type 1
T2DM - Diabetes mellitus type 2
T-cell - T lymphocyte TLR - Toll-like receptor
TIRAP - Toll-interleukin 1 receptor (TIR) domain containing adaptor protein
TNF-a - Tumour necrosis factor a
TRAM - TRIF-related adaptor molecule
Treg - Regulatory T-cell
TRIF - TIR-domain-containing adapter-inducing interferon-p
VDR - Vitamin D receptor VZV - Varicella zoster virus
Introduction
Diabetes mellitus type 1 is an autoimmune disease resulting from the destruction of insulin-producing P-cells within the islets of Langerhans in the pancreas. It has two distinguishable phases - insulitis and overt diabetes (1). The first is characterized by an infiltration of islets' interstitium by macrophages and CD 8+ T-cells (2). The islets of Langerhans undergo apoptosis and the residual P-cell mass is usually decreased by at least 90% at clinical onset of T1DM (3). The second phase is defined by an insufficient insulin production that results in impaired blood glucose regulation, which in turn leads to hyperglycemia. This ultimately causes both acute and chronic complications of disease.
The disease is generally believed to be a result of the interaction of the immune system with environmental and genetic factors (4). Studies conducted in the last two decades have suggested that the role of the humoral arm of the immune system is pivotal in T1DM pathogenesis. However, recent studies show that islet cell antibodies serve as mere markers of P-cell destruction. The crucial role of the cellular arm of the immune system in the development of disease has become more evident only recently (5).
Toll-like receptors
TLRs are pattern-recognition receptors that have changed little in the course of phylogenesis (6). There are 13 human TLRs known today, the first of which was discovered in 1997 (7). TLRs are mainly found on the surface of macrophages and dendritic cells, i.e. the sentinel cells. However, they are also expressed by tissue cells in the central nervous system, the kidneys, and in the liver (8). By recognizing molecular patterns, TLRs found the basis of the innate immune system's function of pathogen recognition. Since molecular patterns eliciting the inflammatory response can have their sources not only in pathogens, but also in an organism's own cells (they are called DAMPs then), TLRs may mediate pathological cell death (9,10).Thus, TLRs are of particular interest in the study of autoimmune disease.
Different TLRs have the ability to recognize a wide variety of exogenous ligands from bacteria (flagellin, glycolipids, lipopeptides, lipoproteins, LTA and LPS), viruses (dsRNA, ssRNA, DNA), fungi (P-glucan), parasites (profilin), as well as endogenous ligands like fibrinogen, heat shock proteins, and HMGB1 (8). Exogenous ligands recognition is enabled by the presence of leucine-rich repeat motifs that facilitate protein to protein interactions. Activated TLRs trigger a signaling cascade that may use the adaptors MyD88 (the most important one), TIRAP, TRAM and TRIF, which results in the activation of NF-kB transcription (11). In fact, only TLR3 uses TRIF and not MyD88 (11,12). It should also be noted that MAP kinases may be activated as a consequence of ligand binding by TLRs. In general, these mechanisms lead to an increase in the microcellular environment concentration of proinflammatory cytokines, antimicrobial peptides, and type I INFs (11).
Vitamin D
Vitamin D is a name used to describe a group of fat-soluble secosteroids. Of these, cholecalciferol (vitamin D3) and ergocalciferol (vitamin D2) play the most important roles in human physiology. It is well known that vitamin D has other functions apart from regulating calcium homeostasis. Vitamin D influences the immune system, the central nervous system, and muscles (13). Therefore, consequences of vitamin D deficiency are not limited to bone disease, i.e. rickets, osteoporosis, and osteomalacia (14). A significant volume of data points towards a role of vitamin D deficiency in autoimmune, neurologic and cardiovascular disease (15). Vitamin D also seems to influence carcinogenesis.
The synthesis of vitamin D begins in the skin, where cholesterol is exposed to ultraviolet B photons. It should be stressed that this natural mechanism of vitamin D production is the most efficient source of vitamin D that the human organism possesses. This is well illustrated by the fact that at most latitudes, sunbathing for approximately 15 minutes between 10 a.m. and 3 p.m. provides sufficient amount of vitamin D (16). On the other hand, there are few nutritional sources of vitamin D, they are not well represented in the Western diet, and they contain insufficient amounts of the vitamin to meet demands. Oily fish, egg yolks, and artificially fortified, highly-processed foods are all examples of nutritional sources of vitamin D (14).
The actual product of biosynthesis of vitamin D in the skin from cholesterol is 7-dehydrocholesterol. It is metabolized to 25-hydroxyvitamin D by the liver. This form of vitamin D is the most commonly used marker of vitamin D status. 25-hydroxyvitamin D is further hydroxylated in the kidneys, yielding 1,25-dihydroxyvitamin D, the most potent form of naturally occurring vitamin D (17).
A commonly used measure of vitamin D deficiency is 25-hydroxyvitamin D serum concentration below 75 nmol/L (17). Although vitamin D deficiency is widespread and seems to be widely prevalent in healthy pediatric and adult populations (18), the recommendations on daily vitamin D intake are conflicting. They most often range from 400 IU daily for infants (United States Institute of Medicine) (19) to 2,000 IU daily for adults over 50 years old (Scientific Advisory Council of Osteoporosis Canada) (20). It was shown that doses as large as 1,500 IU of vitamin D daily may be required to reach vitamin D adequacy. It is also recognized that vitamin D supplementation with doses up to 10,000 IU is safe (21). However, it should be noted that the evidence behind the definition of vitamin D deficiency and the recommendations of its supplementation still leaves many questions unanswered. A recent study by Autier et al. suggests that low 25-hydroxyvitamin D levels in various diseases result from associated chronic inflammation and may not play role in etiopathogenesis (22).
Epidemiology of diabetes mellitus type 1
T1DM constitutes 5-10% of all cases of diabetes (24). According to Diabetes Mondiale Project Group, the incidence of T1DM varies from 0.1/100,000/year in China and Venezuela to 36.5/100,000/year in Finland and 36.8/100,000/year in Sardinia. Twenty-one of thirty-nine European populations have a high or very high incidence of T1DM (10-19.99/100,000/year and >20/100,000/year, respectively). The incidence of T1DM among children increases with age and is highest among 10-14 years old in most populations worldwide (23). EURODIAB data in 17 European countries registered 29,311 new cases of T1DM in children aged under 15 between 1989 and 2003. The overall annual increase in incidence was 3.9% (95% CI 3.6-4.2). The predicted number of new cases in 2020 is 24,400. The incidence among children under 5 years old will double between 2005 and 2020. The prevalence among children younger than 15 years old will rise by 70% in this period (24).
The genetic factors of diabetes mellitus type 1
The cumulative significance of genetic factors is estimated to be as high as 50-65% according to twin studies (25). Approximately 70% of type 1 diabetics carry a HLA risk allele. However, only 3-7% of those carrying such a HLA haplotype will manifest T1DM (26). Individuals positive for HLA class II alleles - HLA-DRB103 (HLA-DR3), HLA-DRB104 (HLA-DR4) and DQB103:02 (HLA-DR8) have the highest risk of developing the disease (27). The link between HLA class II molecules and immune-mediated destruction of the pancreatic islets is still incomplete. The binding of crucial peptides from autoantigens such as GAD, insulinoma-associated 2 antigen, preproinsulin and zinc transporter ZnT8 in the thymus and peripheral organs of the immune system probably play an important role (28). Additionally HLA class I alleles, especially B5701 and B*3906, are independently associated with the disease (29).
Beyond HLA genes, there are also other loci that are linked to T1DM. Current data point towards the following genes: CD69, CTLA4, GLIS3, IL2Ra, IL10, IL19, IL20, IL27, PTPN22, and UBASH3A (3032). There is also an association between T1DM and single-nucleotide polymorphisms in the interferon-induced helicase 1 gene. This gene encodes melanoma differentiation-associated protein 5 -a cytoplasmic sensor for viruses, especially coxsackie B (33).
The environmental factors of diabetes mellitus type 1
Despite the large volume of research, the factor or factors triggering T1DM have not been established thus far. Findings obtained from many studies such as American DAISY (34), German BABYDIAB (35) and Finnish reports (36) are contradictory and do not give a satisfying answer to the T1DM problem. The traditional view of T1DM postulates that an environmental factor causes disease in genetically susceptible individuals, whereas newer theories indicate that the penetrance and the expression of heritable immune aberrations, as well as the progression of initially inherited organ defects, are under the chronic influence of environmental factors (37).
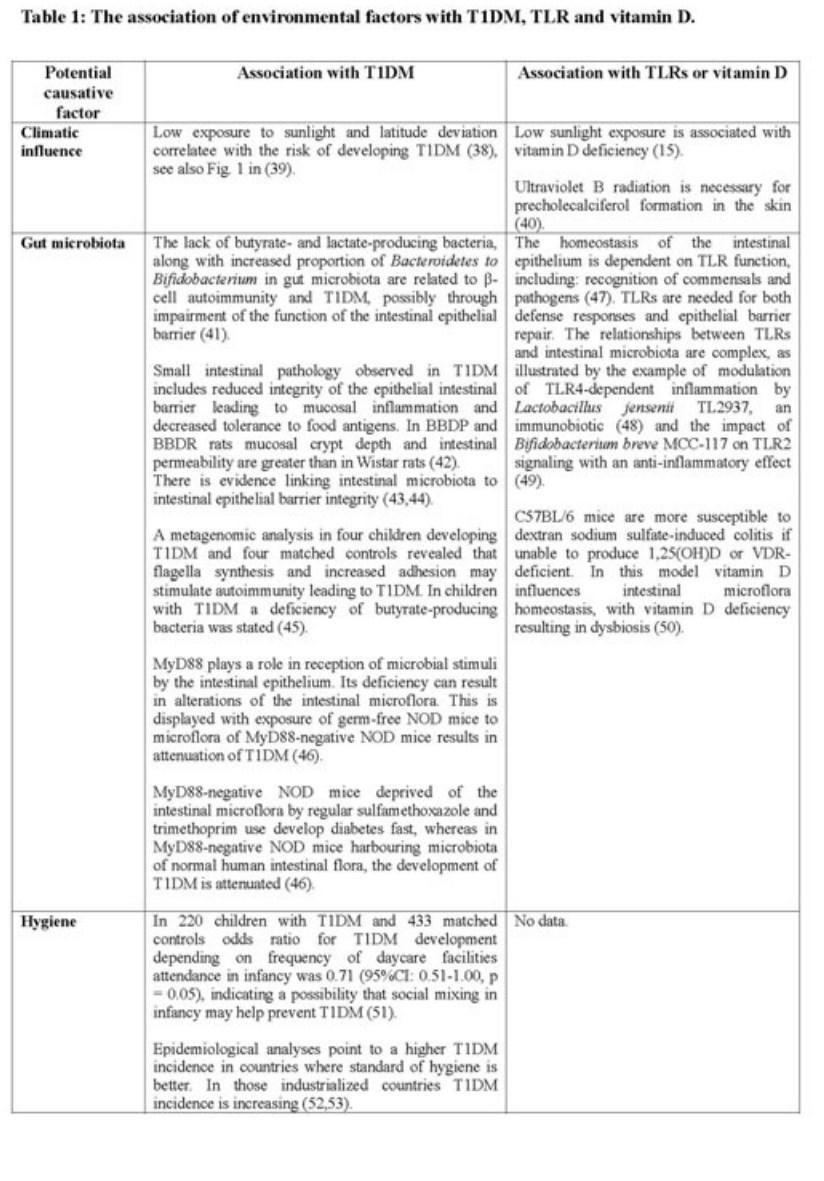
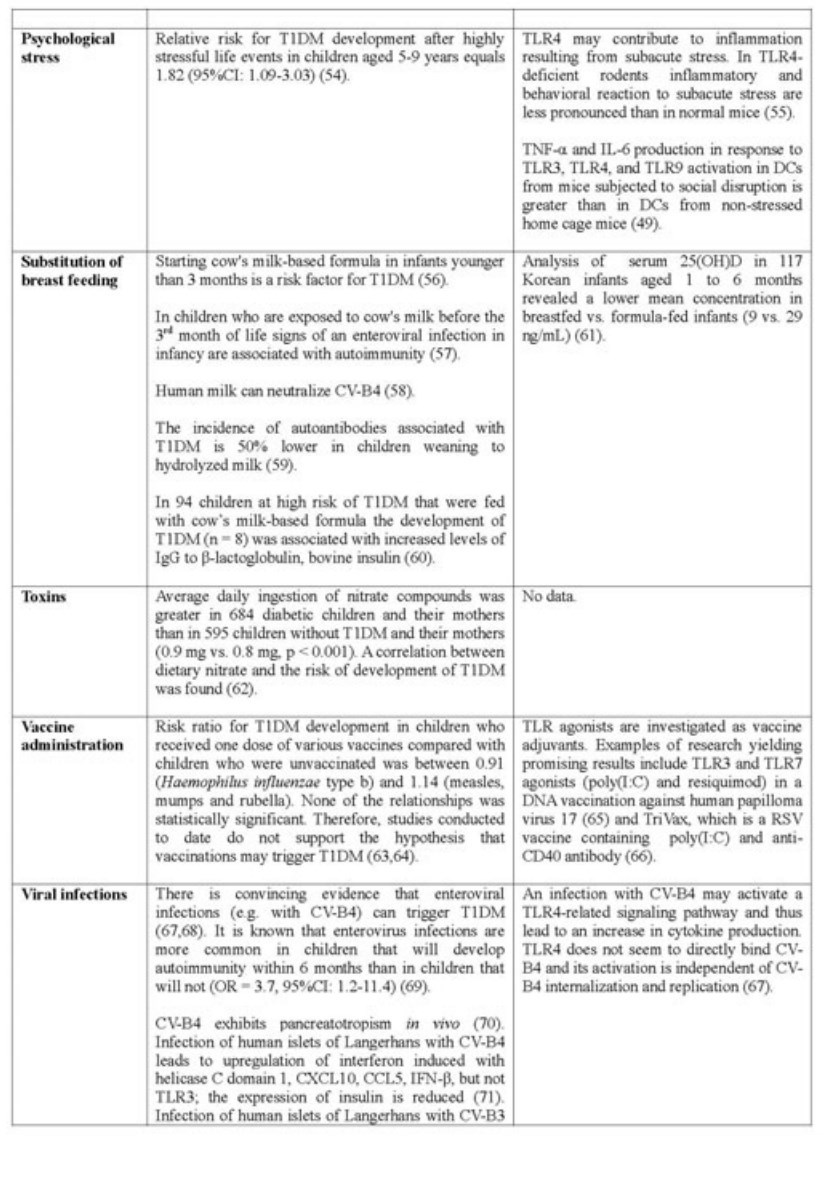
Data concerning most investigated environmental risks in relation to TLRs and vitamin D are summarized in the table below (Table 1). Vitamin D deficiency, as it is presented in Table 2, is considered as an environmental risk factor as well.
Animal models of diabetes mellitus type 1
NOD mice and biobreeding rats are the most popular rodent models in T1DM research. NOD mice
The strain was identified and bred in the late 1970s. NOD mice spontaneously develop T1DM that bears the molecular and clinical features of the disorder in humans, i.e. primary mediation by T lymphocytes and a chronic course. The inflammation of the islets that begins at the age of 4-5 weeks leads to a gradual loss of P-cells and ultimately results in insulin deficiency. Clinical symptoms of T1DM in NOD mice present at the age of 12-30 weeks. It is noteworthy that NOD mice do not require daily administration of insulin and do not present with ketoacidosis. The percentage of NOD mice that develop clinical T1DM is 90% in females and 60% in males. This animal model of T1DM seems to be as complex as T1DM in humans, in addition to its molecular basis also not being fully understood (1,79,80).
Biobreeding rats
The biobreeding rats (BB) were first recognized in the 1970s. There are two inbred strains of BB rats: diabetes-prone (BBDP) and diabetes-resistant (BBDR). In BBDP a T-cell deficiency resulting from a mutation in the gene Ian4 that encodes a mitochondrial protein is at the core of the pathology that usually ensues at the age of 12 weeks. It includes polyuria with polydipsia, hyperglycaemia, weight loss, all linked with the lack of insulin. Unlike in NOD mice, the ketoacidosis is often fatal, and the mice may require insulin for survival. Development of T1DM in BBDR rats, who have normal immunologic phenotype, seems to be dependent on anti-viral antibodies (2,11,79).
As the animal models of T1DM are a result of inbred selection for hyperglycaemia, it is highly probable that not all amplified characteristics of the phenotype are related to the autoimmune processes that underlie T1DM (79).
The immune system in diabetes mellitus type 1
Although the exact mechanism leading to P-cell destruction and T1DM pathogenesis remains unclear, there are theories that seem to integrate the available data well.
In 2001, Mathis et al. proposed that T1DM has its roots in exposure of naive T-cells to islet antigens in lymph nodes. According to this theory, circulating naive T-cells encounter APCs of the pancreatic lymph nodes that carry islet antigens. The sources of the antigens may be diverse, and may include products of cells constituting pancreatic islets and also the remains of apoptotic cells picked up by immature DCs. The T-cells activated after exposure to antigen peptides originating in P-cells may lead to a progressive loss of P-cells. It remains largely unknown in which circumstances the antigens of pancreatic islets could be picked up by the DCs (1).
Lien et al. proposed in 2009 an explanation of mechanisms leading to the development of T1DM in BBDR rats. It concentrates on the role of viruses and TLRs. It is postulated that DCs become activated in response to KRV and TLR ligands. The DCs would upregulate the expression of molecules of class II MHC and chemokines, leading to a proinflammatory state. Activation of signaling pathways linked with TLR9 in response to KRV infection results in inflammation in the lymph nodes of the pancreas. T-cells recruited to those lymph nodes become activated. Simultaneously, regulatory T-cells are systemically downregulated through exposure to cytokines and TLR activation. These events, involving infection and an increase in TLR activity, lead to a release of islet antigens, which are picked up by DCs and presented to T-cells (11). There are also other explanations, which put more stress on the role of an unknown environmental insult. They are based on research conducted with the use of STZ (81).
The "fertile field" hypothesis formulated in 2003 aimed at incorporating the information regarding the roles of various infectious agents in the development of T1DM. The "fertile field" itself is a period of time following a viral infection, during which autoreactive T-cells may expand, resulting in autoimmunity and ultimately T1DM. Therefore, P-cell death is a consequence of interaction between innate susceptibility and an environmental trigger (82). It is noteworthy that P-cells are especially prone to being exposed to pathogenic T-cells as they increase the production of IFN and MHC class I (83). The function of those effector T-cells is not sufficiently attenuated by regulatory T-cells because of their decreased population, altered reactions, and a proinflammatory environment (84). The antigens released from P-cells on their destruction are transferred to the pancreatic lymph nodes by APCs, possibly including immature DCs. Insulin autoantibody production begins after the conversion of B cells to plasma cells. This process occurs in the presence of P-cell antigens. This may in part be due to aberrations in the process of positive and negative T-cell selection in the thymus (85). The autoreactive CD8+ T-cells also play a role in the destruction of P-cells. These processes lead to a second wave of P-cell death. Local inflammation and stress stops insulin production in a large part of the remaining islets. The released antigens further stimulate the autoimmune response, thereby completing the vicious circle of T1DM etiopathogenesis. The proliferation of new pancreatic antigen-specific clones of immune cells is called "epitope spreading". The ensuing inflammation stimulates the proliferation of P-cells, which results in a temporary increase of their mass. The autoreactive process precipitates the onset of T1DM symptoms (82).
The enteroviruses can play a role in the pathogenesis of T1DM through several mechanisms that may yield synergistic effects, e.g. antibody-dependent enhancement of infection, bystander activation of T lymphocytes, IFN-a production by P-cells, molecular mimicry, thymic, pancreatic, and persistent P-cell infection (67).
The roles of toll-like receptors and vitamin D in diabetes mellitus type 1
According to the aforementioned hypotheses and data presented in the Table 2, TLRs play an important role in T1DM pathogenesis. However, the fact that hyperglycemia results in TLR's upregulation should be noted as well (86). The majority of studies indicates that TLR upregulation leads to T1DM development. A few studies described a reduction of risk of T1DM after TLR's early stimulation. This is consistent with the hygiene hypothesis. Data concerning vitamin D's impact on T1DM development is also summarized in Table 2.
Vitamin D and toll-like receptors' effects on the immune system
Information regarding the contradictory effects of TLRs and vitamin D on the immune system, and the associations between them are collected in Table 3.
Conclusions
The current knowledge of TLRs and vitamin D involvement in T1DM points toward new and interesting aspects of autoimmunization and also sheds new light on the vast network of interdependent molecular processes underlying T1DM. The examples of particularly interesting and promising topics related to the role of TLRs in T1DM development are: the normal and pathological course of enteroviral infections, the relationship between the gut microbiota and the immune system, and the impact of substitution of breastfeeding on long-term predisposition to autoimmune disease. It is a matter of discussion whether currently available rodent models of T1DM will be sufficient to answer the arising questions.
Although there is abundant evidence for the role of TLRs and vitamin D in the pathogenesis of T1DM, the exact mechanisms remain elusive. It seems that TLRs exert their influence on the development of T1DM through the modulation of immune responses following P-cell destruction as well as to triggering factors, such as enteroviruses. As such, both TLRs and vitamin D are of particular interest in T1DM research that could potentially pave the way for new clinical interventions. It is noteworthy to mention that such interventions would be effective regardless of the nature of the triggering environmental factor. Supplementation of vitamin D, which is a multidirectional modulator of TLR function, is one such potential intervention, and initial results of trials assessing its efficacy are promising. However, future trials and observational studies are needed to confirm these findings.
References are in PDF
A small portion of the tables in the PDF
PDF is attached at the bottom of this page
See also VitaminDWiki
Search VitaminDWiki for "Toll-like receptors" 201 items as of Oct 2017
Cardiovascular Diseases associated with both Vitamin D and innate immune system (TLR) – Oct 2017
Overview Diabetes and vitamin D contains the following summary
{include}
See also web
- WikiPedia
- They receive their name from their similarity to the protein coded by the toll gene identified in Drosophila in 1985
- The researchers were so surprised that they spontaneously shouted out in German, "Das ist ja toll !" which translates as "That's great!"
- The first reported human toll-like receptor was described by Nomura and colleagues in 1994
- Toll-like receptors are now counted among the key molecules that alert the immune system to the presence of microbial infections.