Major interactions between Vitamin D, Vitamin A, and Influenza
^This PDF to text extraction has the most errors I have every seen. Lost many “f”’s, Many of “The” came out as “Te”, “These” show up as “Tese”, words were strung together etc. etc. Sorry
PDF is attached at the bottom of this page
See also VitaminDWiki
Response to high dose vitamin D is limited by vitamin A - July 2013
Synergism between vitamin A and vitamin D – Masterjohn June 2010
Influenza Mortality not well modeled by vitamin D – Oct 2011
^
Role of Fat-Soluble Vitamins A and D in the Pathogenesis of Influenza:
A New Perspective
Anthony R. Mawson
Department of Health Policy and Management, School of Health Sciences, College of Public Service, Jackson State University, 350 West Woodrow Wilson Avenue, Room 229, Jackson, MS 39213, USA
Correspondence should be addressed to Anthony R. Mawson; amawsn@gmail.com Received 4 April 2012; Accepted 3 May 2012 Academic Editors: M. C. W. Chan, N. Kawai, and Y. Lai
Reduced exposure to solar radiation, leading to a deficiency of vitamin D and hence impaired innate immunity, has been suggested as a trigger for influenza viral replication and as an explanation of seasonal influenza. Although this hypothesis accounts for many unexplained facts about the epidemiology of influenza, gaps remain in understanding the pathogenesis and manifestations of the disease. Several observations suggest a role for vitamin A compounds (retinoids) in the disease. This paper presents a new model of the etiopathogenesis of influenza, suggesting that host resistance and susceptibility depend importantly on the ratio of vitamin D to vitamin A activity. Retinoid concentrations within normal physiological limits appear to inhibit influenza pathogenesis whereas higher background concentrations (i.e., very low vitamin D: A ratios) increase the risk of severe complications of the disease. There is also evidence that influenza-induced or preexisting liver disease, diabetes, and obesity worsen the severity of infection, possibly via liver dysfunction and alterations in retinoid metabolism. The model could be tested by determining the presence of retinoids in the secretions of patients with influenza and by studies of retinoid profiles in patients and controls. Potential strategies for prevention and treatment are discussed.
1. Introduction
"I had a little bird
Its name was Enza
I opened the window
And in flew Enza."
(Children's marching rhyme, 1918).
Influenza is a respiratory viral illness with three main viral subtypes (denoted A, B, and C) that infect and reproduce in human epithelial cells lining the respiratory tract [1]. Of the three subtypes, the most deadly is influenza A, which is associated with annual epidemics and occasional pandemics [2]. Type A has been isolated from humans, birds, pigs, horses, and sea mammals, while types B and C are found only in humans. Influenza A epidemics typically affect the very young and the elderly and have an international distribution. Deaths associated with influenza are due primarily to pneumonia and typically occur in the elderly. Every year from 50,000 to 70,000 influenza-related deaths occur in the United States, exceeding the total number of US lives lost during the Vietnam War. Influenza thus represents an important public health problem [3]. Persons aged 85 years and older are 32 times more likely to die from influenza than those aged 65 to 69 [4]. Annual deaths related to influenza have doubled in the past two decades, mostly involving influenza A viruses.
The appearance of two respiratory viruses in the past decade—severe acute respiratory syndrome coronavirus (SARS-CoV) and avian influenza H5N1 virus—caused great concern due to their associated case fatality rates of over 60%. In particular, increasing outbreaks of highly pathogenic avian H5N1 influenza virus in poultry and its spread to humans have raised the specter of an imminent influenza pandemic. In the absence of effective control or therapeutic measures, new insights on the molecular pathogenesis are needed as a basis for developing new treatments and preventive strategies [5].
In June 2009, cases of flu-like illness in Mexico were reported, associated with a new strain of H1N1 influenza. This was the second of two pandemics involving H1N1 influenza virus, the first being the 1918 pandemic; hence, it was quickly labeled an influenza pandemic. The H1N1 virus strain was thought to have resulted from a reassortment of bird, swine, and human lu viruses, which further combined with a Eurasian pig flu virus, leading it to be called "swine flu," although it was not caused or spread by pigs. By November 2009, over 206 countries had reported laboratory-confirmed cases. The 2009 H1N1 strain preferentially affected young adults. Risk factors for severe disease included obesity, pregnancy, asthma, chronic obstructive pulmonary disease, neurological disorders, and HIV infection. Most cases were mild. By August 2010, contrary to expectation, the number of cases had declined so greatly that the end of the pandemic was officially announced. During the 2010-2011 season, influenza A H3N2 was the dominant serotype, but the 2009 H1N1 continues to co-circulate with H3N2 and B strains [6].
This paper presents a new model of the etiopathogenesis of influenza, suggesting that host resistance and susceptibility to the disease depend importantly on the ratio of vitamin D to vitamin A; reduced exposure to sunlight and/or preexisting vitamin D deficiency simultaneously increase the accumulation, expression, and potential toxicity of endogenous retinoids, and the decreased vitamin D to vitamin A ratio triggers viral activation or increases susceptibility to novel strains of influenza virus.
It is suggested that increased but normal physiological concentrations of retinoid effectively inhibit influenza pathogenesis whereas higher background concentrations (i.e., very low vitamin D: A ratios) worsen it and induce the severe complications of the disease. influenza-induced or preexisting liver disease, diabetes, or obesity may significantly worsen the outcome of infection, possibly via alterations in retinoid metabolism. Methods for reversing the low vitamin D : A ratio could include increasing exposure to solar radiation, dietary restriction, and pharmacological approaches, and all should be investigated for potential use in the prevention and treatment of influenza.
2. Influenza A Virus Infection
Influenza A virus belongs to the Orthomyxoviridae family of RNA viruses that includes influenza A, B, and C. It is an enveloped virus ranging in size from 80 to 120 nm; its genome consists of eight segments of single-stranded negative sense RNA, which encode 10 or 11 proteins, depending on the isolate [7]. The virus has a spherical structure containing RNA material, studded with two surface glycoproteins, hemagglutinin and neuraminidase. Fifteen different types of hemagglutinin (H) and nine types of neuraminidase (N) are recognized. Hemagglutinins are involved in the attachment of viruses to host cell receptors and in the fusion of the viral and cellular membranes, resulting in the release of virion contents into cells [8]. Neuraminidases free newly manufactured virions from the host cell and facilitate virus spread to target cells in airways and facilitate virus release from infected cells [9].
Influenza A viruses are classified according to their unique surface antigens (e.g., H1N1, H3N2). To date, 16 H subtypes (H1-H16) and nine NA subtypes (N1-N9) have been identified among type A influenza viruses [7]. Outbreaks of human influenza infection are associated with changes in the virus, which can be either gradual or sudden and dramatic [1]. In the case of gradual change (termed "antigenic drift"), genetic mutations gradually transform the surface proteins of the virus, primarily hemagglutinins, such that host antibodies increasingly fail to match surface antigens. Tus immunity to a particular strain of virus has limited value in future outbreaks involving different strains, which results in increased susceptibility to infection.
Sudden and major changes in the virus (antigenic shift) are seen mainly in influenza A viruses and involve a rapid transformation of the surface proteins, rendering the virus unrecognizable to host antibodies. Such shits have the potential to cause pandemic infections. In one type of genetic reassortment, gene segments from a prevailing human influenza virus could mingle with an avian influenza virus through an animal intermediary such as pigs. This was thought to have occurred in the 1957 Asian flu and the 1968 Hong Kong flu epidemics. A second type of gene mutation could involve the reassortment of human subtypes within a human host. Thirdly, a pandemic strain could emerge when an avian or mammalian virus becomes infectious to humans as well as capable of person-to-person transmission, as may have occurred in the so-called Spanish flu pandemic of 1918 [1,10].
In recent years the most commonly reported strain has been H5N1, which emerged as an avian pathogen in 1996. This strain led to an outbreak of chicken-influenza in 1997 in Hong Kong, which killed 6/18 people; 5/9 infected adults died compared to 1 of 9 children infected [11]. This strain has circulated in birds and spread widely, leading to sporadic human infections. By March 11, 2009, 411 cases of humans infected with avian influenza H5N1 virus had been reported, with a cumulative case fatality rate of 62% [6].
Only one month later (April 21, 2009), the US Centers for Disease Control and Prevention announced that another pandemic was imminent, involving a new swine influenza virus (H1N1) that was transmitted to and spread among humans, resulting in international disease outbreaks [12]. However, the 2009 H1N1 influenza virus is likely to become extinct unless it mutates or avoids the already high level of global population immunity [13].
The main influenza virus serotypes that have been confirmed in humans, ordered by the number of known human pandemic deaths, are
(i) H1N1—cause of "spanish flu" in 1918 and "swine flu" in 2009,
(ii) H2N2—cause of "Asian Flu",
(iii) H3N2—caused "Hong Kong Flu",
(iv) H5N1—a pandemic threat,
(v) H7N7—has unusual zoonotic potential,
(vi) H1N2—endemic in humans and pigs.
Influenza and the common cold are the most common infectious diseases in human beings. Diagnosis is based on symptoms, and treatments are mainly empirical, since the mechanisms responsible for the disease manifestations are poorly understood compared to knowledge of the disease-causing viruses themselves. The main symptoms of influenza infection usually last 3-5 days and include high fever, chills, rhinitis, headache, myalgia, malaise, extreme fatigue, and cough. Other poorly understood symptoms include sore throat, sneezing, nasal congestion, watery eyes and sinus pain [14]. Cough occasionally progresses to acute respiratory distress, pulmonary infiltrates, and plural effusion. Symptoms are not restricted to the respiratory system and can include multiorgan failure [15], especially if pneumonia or other secondary medical conditions develop, when complications can be fatal (Centers for Disease Control and Prevention. Influenza fact sheet. Available at http://www.cdc.gov/flu/ about/disease/). The features of influenza overlap with other co-circulating respiratory viruses such as respiratory syncytial virus (RSV) and parainfluenza virus. In elderly and debilitated patients with influenza, the disease may present with less prominent respiratory symptoms and only fever, lassitude, and confusion. Although morbidity and mortality associated with both influenza and RSV disproportionately affect the elderly, the 1918 Spanish influenza pandemic caused 20-50 million deaths worldwide, with proportionately higher mortality among young adults [10, 16].
In a report on the cause of death related to pandemic influenza, lung tissue from 58 soldiers who died of influenza at military bases in 1918 and 1919 was examined. Over 2,000 publications during the period 1919 to 1929 were also reviewed, from which 118 autopsy series reports were identified, representing 8,398 individual autopsies conducted in 15 countries. Results showed that most deaths during the pandemic of 1918-1919 were not caused by the influenza virus acting alone but by bacterial pneumonia that followed influenza virus infection. The samples had been preserved in paraffin blocks and were recut and stained for microscopic evaluation. Examination revealed a spectrum of tissue damage ranging from the characteristic features of viral pneumonia and evidence of tissue repair, to those of severe, acute, secondary bacterial pneumonia as the main disease at the time of death. The virus had destroyed the cells lining the bronchial tubes, including ciliated cells, the loss of which made other kinds of cells throughout the respiratory tract vulnerable to attack from bacteria that migrated down the newly created pathway from the nose and throat [17].
Avian influenza A (H5N1) viruses can cause severe disease in humans, characterized by acute lung injury, progressing rapidly to acute respiratory distress syndrome (ARDS), pneumonia, acute renal failure, and multiorgan dysfunction [18]. Extrapulmonary organs infected with human H5N1 virus include the placenta, trachea, intestine, liver, and brain. Increased mortality risk is associated with high viral load and evidence of virus in extrapulmonary tissues, notably liver and brain [19]. Human H5N1 infections are characterized by increased chemokine and cytokine concentrations in macrophages and respiratory epithelia [20]. The primary targets of influenza infection in the lung are respiratory epithelial cells, which produce large amounts of virus that subsequently infect alveolar macrophages [21] and dendritic cells (DCs) [22].
Three specific markers have now been identified that make a particular influenza virus more or less deadly, the presence or absence of which can be determined by examining the virus genome. One such marker is the presence of a coding sequence for PB1-F2, the smallest protein in the influenza virus repertoire. This coding sequence is not found in all human influenza viruses but is consistently present in viruses that are known to be highly virulent in humans and mice. A second marker of pathogenicity is the extent of similarity between the viral hemagglutinin molecules of the new strain and those of other human viruses. Low identity indicates antigenically distinct hemagglutinin structures, suggesting that transmission between humans will not be diminished by a degree of "herd immunity" resulting from exposure to similar viruses. A third molecular marker relevant to the pathogenicity of avian influenza viruses is the polybasic cleavage site, a protease site in the viral hemagglutinin that enables an expanded array of host proteases to activate the hemagglutinin molecule, enabling virus fusion with a host cell [23].
Annual influenza vaccination continues to be an important public health priority, but current vaccines are neither highly immunogenic among elderly persons [24] nor well accepted by the public [25]. Jefferson et al. [26] sought to identify and assess all randomized controlled trials (RCTs) and quasi-RCTs comparing the effects of influenza vaccines with placebo or no intervention in healthy adults. Based on clinical trials that included over 70,000 people, the authors concluded that influenza vaccines modestly reduce influenza symptoms and working days lost but have no effect on pneumonia, other complications, or transmission. They also noted that studies funded from public sources were significantly less likely to report conclusions favorable to the vaccines and that evidence on influenza vaccines was thin and unreliable.
A more recent study [27] assessed the relative reduction in influenza risk from all circulating influenza viruses in the US, based on randomized trials of licensed influenza vaccines (efficacy) as well as reduced risks based on selected observational studies (effectiveness). The data included 17 randomized controlled trials and 14 observational studies and included only laboratory-confirmed cases of influenza.
Pooled efficacy ranged from 59% to 83%, whereas median vaccine effectiveness was 69% (range 60-93). It was concluded that influenza vaccines provide moderate protection against virologically conirmed influenza, but such protection is greatly reduced or absent in some seasons; new vaccines with improved clinical efficacy and effectiveness were needed to reduce influenza-related morbidity and mortality.
There is no cure for influenza, and the molecular basis of pathogenicity is not well understood. Antiviral prophylaxis and treatment with amantadine and rimantadine have been administered in the past, but adverse effects and the rapid development of drug resistance limit their use [28]. Oseltamivir, a neuraminidase inhibitor, is a relatively new treatment option. Stockpiling of the drug is a feature of pandemic preparedness plans [29], but little is known about the efficacy of oseltamivir in human influenza A (H5N1) virus infection. Influenza A (H5N1) virus with an amino acid substitution in neuraminidase conferring high-level resistance to oseltamivir was isolated from two of eight Vietnamese patients treated with oseltamivir who died of the infection, in one case despite early treatment. The presence of detectable virus after completion of treatment was also associated with a poor outcome [30]. Surviving patients had rapid declines in viral load to undetectable levels during treatment. These observations have suggested that resistance can emerge during oseltamivir therapy and may be associated with clinical deterioration; hence, treatment of influenza A (H5N1) virus infection should include additional antiviral agents. A significant need thus exists to understand the pathogenesis of influenza and related conditions such as severe acute respiratory syndrome (SARS) in order to develop effective treatments. SARS was associated with a near pandemic in 2002-2003 and caused 8,096 known infected cases and 774 confirmed human deaths, a case-fatality rate of 9.6% [31]. Research on SARS, a respiratory disease in humans caused by the SARS coronavirus (SARS-CoV), has suggested that symptom worsening is not due to uncontrolled viral replication but to undefined immunologic damage [32]. This leads us to a discussion of a new theory of the pathogenesis of influenza.
3. The Vitamin D Hypothesis of Seasonal Influenza
It has been proposed that seasonal influenza results from vitamin D deficiency due to lack of exposure to solar radiation, which impairs the innate immune system and triggers viral replication [33-35]. The vitamin D deficiency hypothesis accounts for many hitherto puzzling facts about the epidemiology of influenza, but gaps remain in understanding the pathogenesis and manifestations of the disease. Several observations are reviewed in this paper suggesting an important contributory role for retinoids (vitamin A and its congeners) in the pathogenesis, symptoms, and course of influenza infection.
While working as a psychiatrist at a maximum-security hospital, John Cannell screened his patients for vitamin D and found that all had very low levels. This led him to recommend that they take 2000 IU/d of vitamin D, the US "upper limit of tolerability". Several months later an epidemic of influenza broke out at the hospital. Cannell noticed that none of the patients on his own ward developed symptoms, yet sickness was rampant among patients on adjacent wards, despite intermingling between patients and nurses [36]. This observation suggested to Cannell that vitamin D supplementation protected against influenza, an idea consistent with several facts: influenza is a wintertime illness; children with rickets are at increased risk of respiratory infections; and elderly individuals in most countries are more likely to die in winter than in summer months. Cannell and associates [33-35] proposed that influenza is a dormant viral disease that becomes active in response to vitamin D deficiency. Seasonal fluctuations in influenza were explained in terms of annual fluctuations in 25-hydroxy-vitamin D levels due to lack of exposure to sunlight.
The vitamin D deficiency hypothesis similarly explained the following observations:
(i) the appearance of influenza in winter, when vitamin D levels are at their lowest,
(ii) the disappearance ofinfluenza following the summer solstice,
(iii) the increased prevalence of influenza in the tropics and other areas during rainy seasons,
(iv) the inverse association between influenza and outdoor temperature,
(v) the decreased incidence of colds among children exposed to sunlight.
Activated vitamin D,1,25(OH)2D, a steroid hormone, is an immune system modulator that reduces the expression of inflammatory cytokines and increases macrophage function. Vitamin D also stimulates the expression of potent antimicrobial peptides (AMPs), which exist in neutrophils, monocytes, natural killer cells, and epithelial cells of the respiratory tract [37].
Other observations explained by the vitamin D hypothesis are that
(i) volunteers inoculated with live influenza virus in winter were more likely to develop fever and serologic evidence of an immune response than in summer months;
(ii) vitamin D deficiency predisposes children to respiratory infection;
(iii) ultraviolet (UV) radiation reduces the incidence of viral respiratory infections;
(iv) vitamin D supplementation reduces the incidence of respiratory infections in children [33].
The vitamin D deficiency hypothesis accounts for many hitherto unexplained facts about the epidemiology of influenza [38, 39]. Influenza is an allegedly highly infectious viral illness that shows marked seasonal fluctuations, peaking in the winter months and then ending abruptly; it has an obscure serial interval, with a very low secondary attack rate that occurs simultaneously in countries of similar latitude; it spreads very rapidly despite the absence of modern transportation; a high percentage of seronegative volunteers escape illness or experience only a mild illness ater being inoculated with novel influenza virus; and vaccine effectiveness is questionable [40, 41].
Hope-Simpson [39] argued that the epidemiology of influenza was inconsistent with the concept of a highly infectious illness sustained by a continuing chain of transmission from the sick to the well. Although influenza is still considered highly infectious [42], there is surprisingly little evidence to support this assumption. influenza epidemics are often associated with close human interaction, and the first person identified with illness is usually considered the index case. However, the fact that person A becomes sick before person B does not necessarily imply that A infected B. Hope-Simpson suggested that epidemic influenza propagates by a series of transmissions from a small number of symptomless latent carriers temporarily rendered highly contagious by an unknown "seasonal stimulus" that was related to solar radiation and that controlled the seasonality of influenza by rendering human populations susceptible to the disease.
Cannell and his associates [33-35] proposed that this seasonal stimulus is an impaired production ofantimicrobial peptides (AMPs) due to reduced concentrations of 25-hydroxyvitamin D(25(OH)D), following reduced exposure to solar radiation. Support for the suggestion of a marked seasonal decline in vitamin D levels is provided by a prevalence survey of hypovitaminosis D in the British white population [43]. 25-Hydroxyvitamin D[25(OH)D] was measured in 7,437 whites from the 1958 British birth cohort when they were 45 years old. Hypovitaminosis D was highest during the winter and spring; 25(OH)D concentrations <25, <40, and <75nmol/L were found in 15.5%, 46.6%, and 87.1% of participants, respectively, compared to 3.2%, 15.4%, and 60.9%, respectively, during the summer and fall. 25(OH)D concentrations were significantly higher in participants who used vitamin D supplements or oily ish than in those who did not (P < 0.0001 for both). 25(OH)D concentrations < 40 nmol/L were twice as common in the obese as in the nonobese and in Scottish participants, as in those from England and Wales. The prevalence of hypovitaminosis D in the general population was described as "alarmingly high" during the winter and spring, warranting action at a population level.
Seasonal variation in vitamin D levels also occurs around the equator [44], due to sun avoidance [45], rainy seasons, and air pollution. Cannell et al. [34, 35] theorize that epidemic influenza is due to marked variations in the infectivity of infected persons, combined with vitamin D deficiency as the seasonal stimulus. On this basis they propose explanations for a number of hitherto unresolved questions and issues.
(1) Why is influenza both seasonal and ubiquitous, and where is the virus between epidemics? It is postulated that the disease is widely seeded in the population, which explains its ubiquity, while seasonal impairments in innate immunity similarly allow for seasonal epidemics in temperate latitudes and less predictable epidemics in tropical areas. Extensive, out-of-season outbreaks, such as the 1918 pandemic, could arise when "novel antigenic viruses with significantly greater infectivity and virulence overwhelm innate immunity" [34, 35].
(2) Why do epidemics end so abruptly? It is speculated by Cannell et al. [34,35] that this maybe due to the rapid demise of those segments of the population with both impaired innate and adaptive immunity.
(3) Why are influenza epidemics so explosive? Cannell et al. [34,35] suggest that abrupt fall-winter impairment in innate immunity renders a percentage of the non-immune population highly susceptible to background influenza virus.
(4) What accounts for the frequent coincidental timing of epidemics in countries of similar latitude? Tis may be due to simultaneous impairments in innate immunity at similar latitudes resulting from sunlight deprivation.
(5) Why is the serial interval difficult to quantify? The presence of "good transmitters" of the virus as well as vitamin D-induced variations in innate immunity could affect influenza's incubation period and further obscure the serial interval.
(6) Why is the secondary attack rate very low (about 20%); indeed, impossibly low for a highly infectious virus supposedly spreading from sick to well individuals? Because only a subpopulation of the infected (the "good transmitters") are infective. Such individuals cannot yet be identiied.
(7) Why did epidemics in previous ages spread so rapidly despite the lack of modern transportation? influenza is embedded in the population and only erupts when impairments in innate immunity create a susceptible subpopulation, and thus it only appears to be spreading. The occurrence of influenza in large segments of the population seasonally and almost simultaneously may relect the availability of good transmitters. Te disease could actually spread as well, due to the movements of good transmitters.
(8) Why does experimental inoculation of seronegative humans consistently fail to cause illness? Because of variations in the innate immunity of the volunteers, possibly due to variations in 25(OH)D levels.
(9) Why has influenza mortality in the aged not declined with increasing vaccination rates in the past 20 years? Possibly because while vaccination has improved adaptive immunity among the aged, innate immunity among the aged has declined due to public health warnings to avoid sunlight [46]. Cannell et al.'s [34, 35] thesis that vitamin D deficiency is the seasonal stimulus to influenza is supported by the facts that lower respiratory tract infections are not only more frequent in those with low 25(OH)D levels [47] but vitamin D deficiency maybe very common during the 'lu season as well [48].
The basis of the protective effect of vitamin D is said to lie in its ability to stimulate innate immunity and to reduce inflammation [37, 49-51]. Innate immunity differs from adaptive immunity in that the former responds rapidly to microorganisms using genetically encoded effectors, most notably the antimicrobial peptides (AMPs) [52]. As noted, the active form of vitamin D (1,25-dihydroxyvitamin D; 1,25-OH2D) stimulates the expression of AMPs "endogenous antibiotics" in human monocytes, neutrophils, and epithelial cells [37]. AMPs protect epithelial surfaces by destroying the lipoprotein membranes of microbes such as influenza
viruses. If the epithelial mucosal surface barrier is breached, microbes binding to the epithelia promote the expression of inducible AMPs including the defensins and cathelicidin. The defensins inhibit influenza hemagglutinin A-associated carbohydrates [53] and act with cathelicidin as chemoattrac-tants for macrophages and neutrophils [54]. Vitamin D thus enhances the capacity of the epithelium to produce AMPs following exposure to microbes [34, 35]; it also dampens the proinflammatory peptides interferon gamma, TNF-alpha and IL-12 of the adaptive immune system, especially those responsible for acute inflammation "cytokine storms" [55].
Recognition of microbial particles by toll-like receptors (TLRs) induces expression of antimicrobial peptides such as defensins and cathelicidins, which act broadly against microorganisms, including bacteria, fungi, and viruses. Stimulation ofTLRs engages a vitamin D-dependent intracellular circuit that results in the expression of cathelicidin, enhancing the microbicidal capability of the monocyte [56]. Sera from African Americans, who have substantially lower serum vitamin D levels than whites, were inefficient in inducing genetic expression of cathelicidin, but supplementation with vitamin D increased cathelicidin levels to those seen in monocytes from whites. 1,25-OH2D induces expression of cathelicidin and defensin /?2 genes [37], and defensin /?2 has inhibitory effects on adenovirus and HIV-1 [57, 58]. Defensins block viral infection by directly acting on the virion or by affecting the target cell and indirectly interfering with viral infection [58]. One of the defensins (retrocyclin-2) inhibits influenza virus infection by blocking membrane fusion mediated by viral hemagglutinin [53]. These findings have suggested that vitamin D supplementation could prevent colds and influenza.
There is presently little direct or experimental evidence to support the hypothesis that vitamin D might protect against influenza infection. Experiments have not yet been done in cells or mice to evaluate the effect of vitamin D on influenza virus [59].
In one randomized clinical study, 104 postmenopausal African American women were given vitamin D3 (800 IU/d) and 104 were given placebo. After 2 years, the vitamin D3 dose was increased to 50 [ig/d (2000IU) in the active group. Ater 3 years, a total of 34 patients reported symptoms of colds and influenza, eight in the vitamin D3 group versus 26 in the placebo group, a threefold reduced risk (P < 0.002). Te placebo group had cold/influenza symptoms mostly in the winter. Te vitamin D group had symptoms throughout the year while on 20 g/d, but only one subject had a cold/influenza while on 50 g/d. It was concluded that vitamin D supplementation, particularly at higher doses, may protect against the "typical" winter cold and influenza [50] (see Figure 1).
Urashima et al. [60] conducted a randomized, double-blind, placebo-controlled trial comparing the effect of vitamin D3 supplements (1200 IU/d) with placebo on the incidence of seasonal influenza A in schoolchildren, diagnosed with influenza antigen testing with a nasopharyngeal swab specimen. Influenza A occurred in 10.8% of 167 children in the vitamin D3 group compared with 18.6% of 167 in the placebo group (RR: 0.58; 95% CI: 0.34,0.99; P = 0.04), but the Incidence of reported cold and influenza symptoms according to season.
Figure 1 :
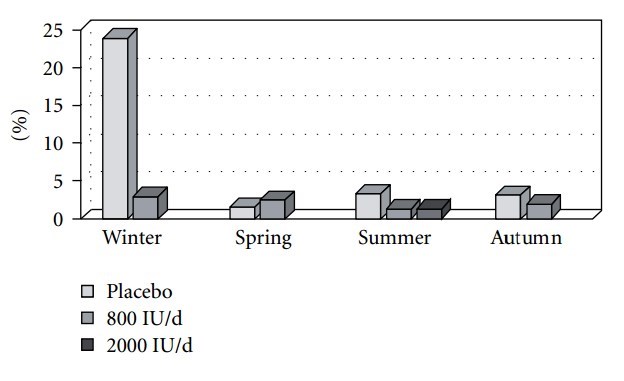
Subjects (» = 104) in placebo group (light shading) reported cold and flu symptoms year-round, with most symptoms in winter. While on 800 IU/d (intermediate shading), 104 test subjects were as likely to get sick in summer as in winter. Only 1 of 104 test subjects had cold and/or influenza symptoms during the final year of the trial, when they took 2,000 IU/d of vitamin D (dark shading; from Cannell et al. [34,35], modified from Aloia and Li-Ng [50] with permission).
protective effect was more marked in children who had not been taking other vitamin D supplements (RR: 0.58; 95% CI: 0.17,0.79; P = 0.006) and who started nursery school after age 3 years (RR: 0.36; 95% CI: 0.17, 0.78; P = 0.005). In children with a previous diagnosis of asthma, attacks of asthma were also significantly reduced in the vitamin D supplement group compared to the placebo group (P = 0.006). Tis study suggests that vitamin D3 supplements during the winter may reduce the incidence of influenza A.
Plasma levels of 25(OH)D are lower in African than in white Americans and are insufficient to stimulate the vitamin D-dependent AMPs, although supplementary 25(OH)D can enhance AMP expression [56]. High melanin concentrations in dark-skinned individuals shield keratinocytes from the UV radiation that generates vitamin D in the skin [61]. Production of vitamin D in skin also diminishes with aging [54]. Hence dark-skinned and aged individuals are at risk of innate immune deficiency, especially in winter. Since vitamin D is mostly obtained from sunlight, vitamin D deficiency is generally higher in winter in aged, dark-skinned, and obese individuals as well as in high northern and southern latitudes [48].
4. Role of Retinoids in the Pathogenesis and Symptoms of Influenza: New Hypothesis
The seasonality and other observations on the epidemiology of influenza A are attributed to population-wide states of impaired innate immunity due to reduced exposure to sunlight during the winter months and a resulting deficiency of vitamin D [33-35]. Cannell's theory postulates that influenza epidemics are the result of a dormant disease that becomes active in response to vitamin D deficiency. Although the vitamin D deficiency hypothesis accounts for many hitherto unexplained facts about the epidemiology of influenza, gaps remain in understanding the pathogenesis, symptoms, and course of influenza infections.
Several observations reviewed in this paper suggest that vitamins A and D have interactive roles in influenza and that retinoids (the collective term for vitamin A and its natural and synthetic congeners) have an independent role in influenza infection and pathogenesis. For instance, solar radiation has opposite effects on vitamins A and D, catabolizing vitamin A but increasing the concentration of vitamin D; the effects of the two vitamins are mutually inhibitory; retinoids regulate airway epithelial cell growth, differentiation, and gene expression; the symptoms of influenza are similar to those of retinoid toxicity; supplementary and/or pharmacological concentrations of retinoids induce influenza-like symptoms; viral activity is regulated in part by retinoids; and retinoids influence the mechanisms that both inhibit and contribute to influenza pathogenesis.
It is hypothesized that reduced sunlight exposure and/or preexisting vitamin D deficiency simultaneously increase the accumulation, expression and potential toxicity of endogenous retinoids (i.e., decrease the vitamin D-to-vitamin A ratio), which trigger viral activation or increase host susceptibility to novel strains of influenza virus. Furthermore, while normal physiological concentrations ofretinoid appear to work with vitamin D to inhibit influenza pathogenesis, higher background concentrations (very low vitamin D: A ratios) worsen it and may induce the severe or lethal complications of the disease. In short, the outcome of influenza infections may depend in part on the ratio or balance between background concentrations of vitamin A and vitamin D. The role of vitamin D and vitamin A in influenza could also, of course, extend to genetic differences in the metabolism and availability of these vitamins (c.f. [62]).
4.1. Retinoids. Vitamin A and its natural and synthetic congeners "retinoids" are mainly dietary-derived fat-soluble signaling molecules that are stored principally in the liver and are essential for normal cellular homeostasis, embryonic development, tissue differentiation, growth, and mucus secretion [63,64]. Retinoic acid (RA), the active form of vitamin A in most cellular differentiation systems, binds to and activates specific retinoid receptors (retinoic acid receptors (RARs); and retinoid X receptors (RXRs)) that regulate the transcription of many target genes [65-67].
The RARs and RXRs are members of the lipophilic steroid/thyroid superfamily of ligand-dependent nuclear transcription factors and include the steroids, retinoids, thyroid hormones, and vitamin D3. Because they can readily diffuse from a source and permeate a target, these lipophilic hormones are potent regulators of development, cell differentiation, and organ physiology [68, 69]. The RARs and RXRs exist as three distinct gene products—a, /?, and y. Upon ligand activation, these receptors function as heterodimeric transcription factors and control the expression of target genes by binding to specific DNA sequences, termed RA response elements (RAREs) [70, 71].
Retinoic acid is produced from free retinol by hydrolysis of retinyl esters stored in the liver and the release of retinol into the circulation and delivery to the target organ tissues bound to retinol-binding protein (RBP). Retinol is first oxidized to retinaldehyde via an alcohol dehydrogenase, and then RA is synthesized from retinaldehyde primarily within the cell microsomes via the enzyme retinaldehyde dehydro-genase. Serum retinol levels remain stable due to a carefully regulated transport system that ensures that the target tissues receive the necessary amounts of retinol despite major ffluctuations in dietary intake [72]. Retinoic acid also influences the action of many transcription factors, for example, repressing the activation of AP-1 by inhibiting the induction of c-Jun and c-fos [73]. RA also regulates other nuclear receptors including the peroxisome proliferator-activated receptors (PPARs), vitamin D receptor, the liver X receptor, and farnesoid X receptor, which heterodimerizes with RXR and regulates the activation of transcription factors, for example, NF-jcB, AP-1, and STAT-1 [67]. RA exerts opposing effects on cell growth via the alternative activation of RARs versus PPARjSS [74].
Low concentrations of retinoic acid are essential growth factors for certain types of cells, but higher concentrations inhibit cell growth and are cytotoxic, mutagenic, and ter-atogenic. Exogenous vitamin A toxicity can occur due to excessive dietary consumption or from treatment with reti-noids. Although vitamin A toxicity from provitamin A plant carotenoid sources has never been reported, the absorption and hepatic storage of preformed vitamin A from animal foods, fortiied foods, and supplements in the form ofretinyl esters can result in hypervitaminosis A. An endogenous form of retinoid intoxication can also occur naturally during cholestasis, when vitamin A metabolites are reluxed into the circulation from the liver in bile acids [75]. A variety of environmental factors can interact with endogenous sources of vitamin A to induce localized forms of retinoid toxicity or overexpression, as reviewed in this paper.
Retinyl esters in serum, normally <0.2 mol/L in the fasting state, increase significantly after a large vitamin A-containing meal, after which they are converted to retinol and stored in the liver. Retinol binds to RBP and is transported to the target tissues. Vitamin A toxicity is generally associated with increased levels of retinyl esters circulating with plasma lipoproteins unbound to RBP. Retinyl esters react more randomly with cell membranes than the physiologically sequestered RBP and hence are a major form of vitamin A toxicity. Fasting retinyl ester concentrations >10% of total circulating vitamin A (retinol plus esters) are considered a biomarker for toxicity [76]. An acute increase in the concentration of other retinoids, for example, retinoic acid, a 40-fold more potent teratogen than retinol [77] occurs after ingesting a large amount of vitamin A. Retinoic acid and other acidic retinoids are much more biologically active and hence more toxic than retinol. However, the precise ranges of serum retinoic acid associated with symptomatic acute or chronic vitamin A toxicity are not well defined. Treatment with 13-cis-RA at 30mg/kg/d raises circulating levels of retinoic acid from a physiological range of about 1-2 ng/mL to > 10 ng/mL and occasionally to as high as 70 ng/mL [78].
Serum retinol concentrations (normally 1-3 mol/L) do not reflect hepatic vitamin A concentrations over a wide range of liver values, since the secreted RBP is under homeostatic control. Thus serum retinol concentrations vary little despite major alterations in vitamin A intake. Case reports of hypervitaminosis A often show serum retinol concentrations within normal limits, indicating that serum retinol is not a valid measure of vitamin A status during toxicity [76, 79].
Research on vitamin A toxicity has been carried out mostly in animals, but observational studies suggest that >75% of people in developed countries routinely consume more than the recommended dietary allowance (RDA) for vitamin A [80]. Children are particularly sensitive to vitamin A, since toxicity can be induced with daily intakes of 1500 IU/kg body weight [81]. Prospective studies have identified an association between preformed vitamin A intake and hip fracture or osteoporosis ([76], for review). The vitamin A dietary intakes related to bone disease were low (1500 RE): half the amount usually associated with the risk of toxicity and lower than the highest amount thought to pose no risk of adverse health effects in the general population, that is, the tolerable upper level of intake (3000 RE). These observations suggest that intakes much lower than the amount conventionally thought to lead to toxicity (10 times the RDA) may increase the risk of osteoporosis, that is, about twice the RDA for adult females (700 RE).
As for more general effects on health, vitamin A is considered an antioxidant vitamin and supplements are widely available as measures to prevent disease. In a Cochrane Review of the effect of antioxidant supplements (including vitamin A) on mortality, based on randomized trials, Bje-lakovic et al. [82] reviewed all randomized trials involving adults consuming beta-carotene, vitamins A, C, E, and selenium either singly or combined. Tey included 68 randomized trials with 232,606 participants. When all trials of antioxidant supplements were pooled together, there was no significant effect on mortality (RR: 1.02; 95% CI: 0.98-1.06). However, multivariate regression analyses showed that, in the better designed "low bias" trials, beta-carotene was associated with a significant 7% increased risk, vitamin A with a 16% risk, and vitamin E with a 4% increased risk of mortality, whereas vitamin C and selenium had no significant effect on mortality. Te authors concluded that treatment with beta-carotene, vitamin A, and vitamin E may increase mortality.
Several lines of evidence support the proposed model, as summarized below.
(i) Vitamin A is sensitive to photooxidation.
(ii) Vitamins A and D are inversely associated in that vitamin A can inhibit the actions of vitamin D and vice versa.
(iii) Retinoic acid, the major metabolite ofretinol, plays an essential role in the regulation of airway epithelial cell growth, differentiation, and gene expression.
(iv) The symptoms of influenza are mimicked by retinoid toxicity, for example, the retinoic acid syndrome, induced by the use of synthetic retinoids for the treatment of acute promyelocytic leukemia.
(v) Supplementary vitamin A can induce influenza-like symptoms.
(vi) Viral activity in general is regulated by retinoids.
(vii) Retinoids influence the factors and mechanisms that both inhibit and contribute to influenza pathogenesis.
5. Solar Radiation Photooxidizes Vitamin A
Solar radiation increases vitamin D levels, as noted above. On the other hand, vitamin A is sensitive to photooxidation and may be destroyed by it. Two forms of vitamin A are found in human skin: all-trans-retinol (A:) and 3-dehydrotretinol (A2). Dermal retinol is derived partly from the adjacent subcutis, which contains 10-20 times more vitamin A than both skin and blood [83]. 3-Dehydroretinol occurs mainly in the epidermis and may be a metabolite of retinol [84]. Small amounts of retinoic acid are also found in human skin and represent a mixture of 13-cis/trans isomers of retinoic acid. In Caucasian human skin, levels of retinyl esters and retinol are 3.5 and 5.0 times higher, respectively, in the epidermis than in the dermis [85].
As an intrinsic modulator of proliferation and differentiation in human epidermis, vitamin A may be destroyed by ultraviolet radiation (UVR) impinging on the skin. The solar spectrum of UVR (290 to 400 nm) is commonly divided into three bands, from longer to shorter wavelength regions: UVA (320-400 nm), UVB (290-320 nm), and UVC (200-290 nm). UVB and UVA radiation are the principal wavelengths causing sunburn [86]. The absorption maxima of most retinoids range from 280 to over 400 nm [87]. From 35% to 50% of incident ultraviolet-A radiation (UVA) is transmitted through Caucasian epidermis [88] and absorbed directly by blood in the capillaries of the papillary dermis. UVA radiation produces histologic changes in skin at greater depths than the shorter UVB or UVC wavelengths [89]. Ultraviolet radiation has a biphasic effect on human blood vitamin A levels [90]: irst, an increase, which peaks at 7 hours following irradiation, and then a fall 24-48 hours later. Tang et al. [85] investigated the effect of sunlight on retinyl esters and retinol in human skin, blood, and cultured keratinocytes. Sunlight irradiation led to a significant reduction in epidermal retinyl esters in Caucasian skin in both summer and winter, whereas epidermal and dermal retinol and dermal retinyl esters were affected to a lesser extent. When serum from volunteers who had taken a large dose of retinyl palmitate to elevate serum retinyl esters was exposed to sunlight, the serum retinyl esters disappeared ater 10 minutes of exposure. Andersson et al. [91] studied endogenous retinoid concentrations and metabolism in cultured human keratinocytes and melanocytes exposed to UVR. Before UVR the retinoid content was similar in keratinocytes and melanocytes. In both cell types, UVR (i.e., UVA 360mJ/cm2 plus UVB 140 mJ/cm2) instantaneously reduced the concentration of retinol by about 50% and that of 3,4-didehydroretinol by about 20%. The uptake of retinol was threefold higher and that of retinoic acid was tenfold higher in the melanocytes, and in both types of irradiated cells the accumulation of the biologically most active metabolite, all-trans retinoic acid, was about 60% higher than in control cells. Retinoid concentrations returned to normal within 12 days after irradiation. The metabolism of retinoic acid was reduced, especially in irradiated keratinocytes, which may have contributed to the restoration of retinoid levels ater UV exposure. These observations point to a complex pattern and sequence of changes in retinoid metabolism following exposure to solar radiation.
6. Vitamins A and D Are Inversely Related
The fat-soluble vitamins A, D, E, and K normally work interactively together. For instance, earlier suggestions that the function of vitamin D requires vitamin A [92] now find support in the important observation that the RXR ligand 9-cis-retinoic acid (9-cis-RA), a hormonally active form of vitamin A, potentiates vitamin D-dependent gene expression and thus has a role in vitamin D signaling that was previously unknown. It was found that when 9-cis-RA acid was unavailable, vitamin D could only bind weakly to DNA and exerted only a small effect on gene expression; conversely, the presence of 9-cis-RA conferred significant agonistic activity to a vitamin D receptor ligand with very low agonistic activity and also increased the differentiation of colon cancer cells by vitamin D. Most remarkably, the addition of 9-cis-RA restored the functioning of a mutant (defective) vitamin D receptor present in a hereditary form of rickets that cannot normally be cured by vitamin D [93].
On the other hand, the evidence suggests that in the event of major perturbations in either vitamin A or D, due to dietary or other factors, there is an inverse, mutually inhibitory relationship between them in that vitamins A and D counterbalance their potentially toxic individual effects [94]. For instance, while vitamin A can reduce the toxicity of vitamin D (e.g., [92, 95]), vitamin D can also interact inversely with vitamin A and reduce the toxicity of vitamin A. Although little is known about the effect of reduced sunlight exposure and/or deficient vitamin D levels on vitamin A metabolism, even small to moderate doses of vitamin D in chickens reduce liver vitamin A stores and lower the level of vitamin A in blood [96]. Exposure of chickens to UV light (which produces vitamin D) likewise reduces liver stores and blood levels of retinol [97]. In humans, concomitant supplementation with vitamin D greatly increases the dose of vitamin A required to cause toxicity; for instance, Myhre et al. [98] found that the median dose for inducing vitamin A toxicity was >2,300 IU/kg of body weight per day higher when vitamin D was added to the diet. For a hypothetical 75 kg person representing the median, vitamin D supplementation would have allowed an additional 175,000 IU vitamin A/day before toxicity symptoms were likely to be reported. In the Nurses' Health Study, a positive association was found between retinol intake and fracture risk in that vitamin D intake increased as retinol intake increased, but at a lower rate. In a multivariate analysis controlling for many factors, vitamin D was found to be protective against retinol-associated risks of fracture [99].
The potential toxicity of high intakes of vitamin A may thus depend in part on the amount of vitamin D consumed. On the other hand, vitamin D deficiency would be expected to increase the potential toxicity of vitamin A. Indeed, consumption of preformed retinol, even in the usual amounts consumed in the United States in the forms of multivitamins, may cause osteoporotic bone changes in adults with low vitamin D concentrations [76]. Scandinavian countries, which have the highest fracture rates in Europe and even worldwide [100], have higher average intakes of vitamin A and are also at far higher latitudes (60 degrees), where vitamin D "winters" (periods of time in which vitamin D cannot be produced by the action of sunlight on the skin) are longer and vitamin D is less available from the sun, even in the months when sunlight is present.
With regard to the impact of vitamin A on vitamin D, in humans the amount of vitamin A in a single serving of liver inhibits the rise in serum calcium induced by vitamin D [101]. Retinoic acid can antagonize the action of vitamin D and its active metabolite 1,25-dihydroxycholecalciferol in rats [102]. A high intake of retinol also completely abolished the protective effect of vitamin D on distal colorectal adenoma, women in the highest quintile of vitamin D intake ingested about 10,000 IU/day of retinol, and there was a strong correlation overall between dietary intakes of vitamins A and D [103].
Sunlight in the ultraviolet (UV-B) spectrum converts a cholesterol precursor, 7-dehydrocholesterol, into the activated form of vitamin D (calcitriol). Thirty minutes of whole-body exposure of pale skin to sunlight with clothing or sunscreen can result in the synthesis of from 10,000 to 20,000 IU of vitamin D. Further radiation converts excess vitamin D in the skin into inactive metabolites. Melanin pigment also accumulates in skin, thereby decreasing the production of vitamin D [104]. On this basis it has been suggested that, in order to maintain serum levels of 25(OH)D at the optimal range of 50 ng/mL during a vitamin D "winter", 4,000 IU vitamin D should be consumed per day [105].
Possible Interactive Roles of Vitamins A and D in the Seasonality of Influenza.
The marked peak in influenza infections in the cold winter months in temperate regions [3] may be explained on the present hypothesis as follows: the seasonal pattern of influenza peaks in the winter months and troughs in the summer may be due not only to seasonal changes in vitamin D availability and its effects on vitamin A but also to the simultaneous influence of solar radiation on vitamin A metabolism, together with seasonal changes in ambient temperature and its effect on vitamin A metabolism.
Vitamin A is affected by changes in temperature as well as light; both light and warm temperatures cause vitamin A to be catabolized. In one study, twin calves were fed 6 mg carotene daily per 100 lb body weight and subjected to high and low ambient temperatures. High temperatures increased liver vitamin A utilization; moreover, identical twin calves exposed to solar radiation for 38 days lost more hepatic vitamin A than their cotwins in the shade [106]. Thus, rising temperatures and greater sunlight in the summer months could catabolize tissue concentrations of vitamin A to such a degree that it would prevent influenza viruses from making use of it to replicate. Conversely, the seasonal increase in influenza during the winter months may occur partly from the fact that vitamin A remains available for the virus to replicate in cooler temperatures.
In summary, reduced solar radiation and cooler temperatures during the winter months may serve at once to reduce vitamin D reserves and to increase the accumulation and hence potential toxicity of vitamin A, that is, to lower the vitamin D: A ratio. Here it is proposed that the increased endogenous concentrations of vitamin A in turn interact with and activate the influenza virus, resulting in the signs and symptoms of infection via molecular mechanisms that are discussed below. If this hypothesis is correct, not only reduced solar radiation and vitamin D deficiency but also vitamin A supplementation (and/or conditions associated with high preexisting retinoid concentrations) could increase susceptibility to influenza, especially in a situation of reduced sunlight exposure and/or vitamin D deficiency. At the same time—given the interaction between vitamins A and D—vitamin D supplementation may protect against influenza, partly by inhibiting the expression of vitamin A. This line of reasoning suggests that seasonal and possibly pandemic influenza could be prevented and treated by methods to increase the vitamin D : A ratio.
7. Role of Retinoids in Affected Tissues
Influenza infection primarily but not exclusively affects the respiratory system. Vitamin A and its active metabolites are likewise importantly involved in the growth and differentiation of mucosa-associated airway epithelia. Delivery of retinol via the bloodstream to target cells ensures a sufficient cellular supply. Vitamin A is stored in the target tissues as retinyl esters, which provide an additional source of the vitamin [107]. Retinoic acid, the major biologically active metabolite of retinol, plays an essential role in the regulation of airway epithelial cell growth, differentiation, and gene expression. Levels of retinoid binding proteins, the RA receptors, and RA synthesizing enzymes all peak post-natally. Retinoic acid is also required throughout life for the maintenance of lung alveoli, and a deficiency leads to a loss of alveoli and to features of emphysema. Exogenous retinoic acid has been reported to induce alveolar regeneration in a rat model of experimental emphysema, and an inhibitor of retinoic acid synthesis, disuliram, disrupts alveologenesis [108].
Using cDNA icroarray, Di et al. [109] identified a clone, DD4, that contains the cDNA of a novel gene, spurt (secretory protein in upper respiratory tracts), which is significantly induced by retinoic acid in primary cultured human tracheobronchial epithelia and is located on chromosome 20q11.21. Spurt mRNA is expressed at high levels in human nasal, tracheal, and lung tissues and secretory cell types and is present in clinical sputum and tissue samples.
8. The Symptoms of Influenza A Infection Are Similar to Those of Hypervitaminosis A
As noted, the clinical spectrum of influenza A infection, including avian influenza H5N1, is not restricted to the lung and can range from mild influenza-like illness to severe pneumonia, acute respiratory distress syndrome (ARDS), acute lung injury (ALI), and multiorgan failure [15]. Fever, rhinitis, myalgia, malaise, headache, cough, dyspnea, sore throat, and fatigue are the main presenting symptoms. Complications include pneumonia, bronchitis, or sinusitis, and rarely encephalitis, transverse myelitis, Reye syndrome, myocarditis, or pericarditis [110]. Additional features associated with severe disease and mortalityinclude diarrhea, vomiting, abdominal pain with widespread viral dissemination, and cardiovascular events (stroke, myocardial infarction) [5]. It has also become clear that influenza virus can cause severe hepatitis and liver damage; indeed, screening with biochemical liver tests is recommended in severe influenza infection [111-113].
The retinoic acid syndrome is a potentially life-threatening complication seen in patients with acute promyelocytic leukemia (APL) treated with all-trans-retinoic acid (ATRA). The syndrome is similar to that of ARDS and commonly includes fever, headache, acute respiratory, pleural or peri-cardial effusions; pulmonary edema, and infiltrates requiring mechanical ventilation in about 25% of patients; eosinophilia, marked basophilia and hyperhistaminemia, granulomatous proliferation, alterations in weight, peripheral edema, thromboembolic events, episodic hypotension, nausea, vomiting, and acute renal failure requiring hemodialysis in some patients. Specific influenza-like symptoms include muscle aches, fever, chills, and fatigue [114, 115].
In an unusual case report, the symptoms of influenza A infection were described as being perfectly mimicked by the retinoic acid syndrome [116]. A 47-year-old man was hospitalized for typical APL and treated with ATRA and chemotherapy. On day 3 the patient developed fever and acute respiratory distress and was admitted to the critical care unit. ATRA was stopped since the diagnosis of retinoic acid syndrome was suspected. Bronchoalveolar lavage and immunoluorescence examination showed the presence of influenza A virus, which was confirmed by the rise of specific antibody levels in sera obtained during the acute illness and 3 weeks later. This case report shows that an infectious disease—influenza A infection—can perfectly mimic the retinoic acid syndrome and suggests that endogenous sources of retinoic acid could contribute to influenza and its sequelae.
Headache, a common symptom of influenza [117], is also a major feature of retinoid toxicity [118]. Conjunctivitis and photophobia are also common during acute seasonal influenza infection, especially in avian influenza A infections in humans [119]. An oculorespiratory syndrome (ORS) consisting ofred eyes, photophobia, blurred vision, palpebral edema, ocular pain and itching, and conjunctival secretions is reported after influenza vaccination [120]. A similar pattern of ocular side effects has been described in diet-induced hypervitaminosis A and secondary to isotretinoin use. In a review of 1,741 spontaneous case reports, as well as data from the Drug Safety Section of Roche Pharmaceuticals and the world literature, adverse ocular reactions classified as "certain" to have been associated with isotretinoin use included photophobia, abnormal meibomian gland secretion, blepharoconjunctivitis, corneal opacities, decreased dark adaptation, decreased tolerance to contact lens, decreased vision, increased tear osmolarity, keratitis, meibomian gland atrophy, myopia, ocular discomfort, and ocular sicca [121]. Similarities between the features of hypervitaminosis A and influenza infection are shown in Table 1.
9. Supplementary Vitamin A Can Induce Influenza-Like Symptoms
It has been recognized for decades that vitamin A deficiency is associated with increased susceptibility to most infections and with defects in the innate and adaptive immune systems [67]. The traditional view of vitamin A as an "anti-infective" vitamin was based partly on earlier studies in which vitamin A—in cod liver oil (CLO)—was successful in preventing infection [122]. Since earlier preparations of CLO contained higher amounts of vitamin D in proportion to vitamin A than do currently available preparations, possibly due to modern deodorization procedures, which remove vitamin D, it has been suggested by Cannell etal. [34,35] that the anti-infective properties of CLO were partly or wholly due to vitamin A.
Consistent with Cannell's hypothesis, vitamin A supplementation has not been shown to improve recovery during acute pneumonia in most human clinical trials. In a double-blind, placebo-controlled trial of vitamin A supplementation on childhood morbidity in Haiti, 11,124 children ages 6-83 months were sequentially assigned by household units to receive either a capsule containing 200,000 IU of vitamin A and 40.6 mg vitamin E or a capsule containing only 40.6 mg vitamin E (placebo) every 4 months. Indicators of childhood morbidity were studied 2-8 weeks after each administration of vitamin A and placebo capsules. At 2 weeks after supplementation the vitamin A group had an increased prevalence of all symptoms and signs of childhood morbidity, including diarrhea, rhinitis, cold/flu symptoms, cough, and rapid breathing. The risk of morbidity was highest 8-17 weeks after receiving the megadose of vitamin A. The study showed an increased 2-week prevalence of diarrhoea and the symptoms of respiratory infections after vitamin A supplementation, although mortality rates of the 2 groups were similar [123]. A meta-analysis of vitamin A supplementation trials concluded that when given alone, vitamin A slightly increased the incidence of respiratory tract infections [124].
Stephensen et al. [125] conducted a randomized, double-blind, placebo-controlled clinical trial to test the hypothesis that high-dose vitamin A supplements would enhance recovery of children ages 3 months to 10 years (N = 95) hospitalized with pneumonia in Lima, Peru. Children <1 year of age received 100,000 IU of water-miscible vitamin A on admission to the hospital and an additional 50,000 IU the next day; children >1 year of age received 200,000 IU on admission and 100,000 IU the next day.
Table 1: Similarities between the signs and symptoms of influenza infection and of hypervitaminosis A (sources: references cited in the text).
Contrary to expectation, by day 3 the children receiving vitamin A had lower blood oxygen saturation (the mean difference was 1.1%), higher prevalence rates of retractions (37% in the vitamin A group, 15% in the placebo group), auscultatory evidence of consolidation (28% in the vitamin A group, 17% in the placebo group), and were more likely to require supplemental oxygen (21 % in the vitamin A group, 8% in the placebo group) compared to children in the placebo group. Adjustment for baseline severity of disease and nutritional status did not alter the association of vitamin A with increased clinical severity; there was no difference in duration of hospitalization or in chest X-ray changes 14 days after admission; no deaths occurred. The authors concluded that high-dose vitamin A supplements caused modest adverse effects in children recovering from pneumonia and should not be used therapeutically in such patients in the absence of clinical evidence of vitamin A deficiency or concurrent measles infection.
Cui et al. [126] tested the hypothesis that high vitamin A intake would decrease the production of T-helper type-1 (T1) cytokines and inhibit antiviral responses and thereby impair recovery from viral respiratory infections. Tree interventions were tested on patients with viral pneumonia: (1) a high-level vitamin A (250,000 IU/kg diet or 75,000 retinol equivalents (RE)/kg), (2) control diet (4000 IU/kg diet or 1200 RE/kg) given before and during infection, and (3) initiating the high level diet upon infection to simulate the adjuvant therapy used in clinical trials. No difference was seen among the interventions in severity of disease in terms of weight loss, lung virus titers, and survival. However, the high-level diet group (and that in which vitamin A was increased at the time of infection) had greafter salivary immunoglobulin (Ig)A responses than did the control group. In contrast, the serum IgG response was higher in the control group than in the high-level group, although it did not differ from the group in which the diet was changed upon infection.
The production of interferon-gamma (IFN-gamma), aThl cytokine, was significantly lower in the high-level diet group compared with the control group, whereas the production of interleukin-10 (IL-10), a Th2 cytokine, was higher with the high-level diet than that with the control. It was suggested that the observed change in the Th1/Th2 pattern was insufficient to affect recovery from viral pneumonia but may have accounted for the increased IgA and decreased IgG responses seen with the high-level vitamin A diet. These data were interpreted as reinforcing the lack of utility of vitamin A in treating acute pneumonia in children, consistent with other evidence that high-dose vitamin A supplements enhance T2-mediated immune responses, which have been found beneficial in extracellular bacterial and parasitic infections and IgA-mediated responses to mucosal infections.
Other evidence suggests that supplementary vitamin A maybe unhelpful and indeed harmful in respiratory illnesses, including influenza and pneumonia. Influenza is commonly associated with lower respiratory tract infections (LTRIs) in children. In a review of the literature on the effectiveness and safety of vitamin A for preventing LRTIs in children up to seven years of age, several databases were searched for randomised controlled trials [127]. Most studies found no significant effect of vitamin A on the incidence of acute LRTI or symptom prevalence. Vitamin A supplementation was associated with an increased incidence of acute LRTI in one study and with an increase in cough and fever and increased symptoms of cough and rapid breathing in two others; three reported no differences and no protective effect of vitamin A; two studies reported that vitamin A significantly reduced the incidence of acute LRTI with children with poor nutritional status or weight but increased LRTI in normal children. Te authors concluded that vitamin A supplements should not be given to all children to prevent acute LRTIs but appeared to beneit children with low serum retinol or poor nutritional status.
In another meta-analysis of the literature on the effectiveness of supplementary vitamin A for infants and children diagnosed with nonmeasles pneumonia, the authors reviewed parallel-arm, randomised and quasi-randomised controlled trials in which children younger than 15 years old with nonmeasles pneumonia were treated with adjunctive vitamin A [128]. Based on five trials involving 1,453 infants and children, no significant reduction was found in deaths from pneumonia in children treated with vitamin A compared to untreated controls. There was also no effect on hospital length of stay, the results of chest X-rays, and symptoms of vomiting, diarrhea, or irritability. Tere was, however, an apparent effect on bulging of the fontanelle (OR: 8.25; 95% CI: 0.44-155.37). Disease severity after supplementary high-dose vitamin A was also significantly worse in children who received vitamin A compared with placebo; on the other hand, low-dose vitamin A was associated with a significant reduction in the recurrence rate of bronchopneumonia (OR: 0.12; 95% CI: 0.03-0.46). This meta-analysis failed to show a significant reduction in mortality and measures of morbidity, and there was no effect on the clinical course of pneumonia in children with nonmeasles pneumonia following vitamin A supplementation.
10. Viral Activity Is Regulated in Part by Retinoids
Consistent with the role of retinoic acid in cell growth and differentiation, viral growth is also regulated in part by vitamin A [129]. A number of virus infections are known to be influenced in complex ways by retinoids. For instance, when different cell lines are infected with human cytomegalovirus (hCMV), exposure of the cells to retinoic acid (RA) enhances viral gene expression and susceptibility to infection [129]. RA also reactivates CMV expression in latently infected glioblastoma cells in tissue culture [130]. Reactivation of latent virus is believed to result from a signal transduction event that induces immediate-early (IE) gene transcription, a crucial viral control element. Ghazal et al. [131] have shown that the major IE promoter of hCMV is activated by physiological levels of RA in human embryonal carcinoma cells (i.e., functions as a retinoic acid response element or RARE) and is a specific target site for the direct interaction of nuclear receptor proteins for RA. These findings suggest that RA is a potential modulator of hCMV pathogenesis.
Murine CMV (mCMV) is also susceptible to regulation by natural and synthetic retinoids at different levels. In tissue culture cells, the major IE enhancer can be activated by RA via multiple RA-responsive elements (RAREs) that bind RXR-RAR heterodimers. Viral growth was dramatically increased following RA treatment of infected tissue culture cells. RAR activation was required to mediate the response of mCMV to RA and selectively promoted viral growth; moreover, the stimulatory effects of RA on enhancer activity and viral growth were prevented by treatment with an RAR-specific antagonist. Oral administration of RA to infected mice worsened an acute infection by mCMV, whereas an RAR-antagonist, also administered orally, protected against the adverse effects of RA in mCMV infection [132].
Human immune deficiency virus (HIV-1) expression in macrophages is similarly enhanced by RA, and retinoid signals are mediated by RARs and RXRs that bind to specific RA response elements (RAREs) in the promoter region of the susceptible genes [133]. A RARE in the long terminal repeat (LTR) region allows activation of the HIV-1 LTR. An RAR-antagonist also strongly inhibited retinoid-induced activation of the HIV-1 RARE [134]. The observation that treatment with an RAR antagonist protects against viral infection induced by RA suggests the possibility that RAR antagonists could be therapeutically useful in viral infections other than CMV.
Noting that retinoic acid (RA) induces epithelial cell differentiation and that the conversion ofretinol to RA requires retinol dehydrogenase enzymes, Jones et al. [135] have shown that gastric carcinoma cells containing a transmissible form of Epstein-Barr virus (EBV) have enhanced expression of a gene (DHRS9) that encodes an enzyme that mediates conversion of retinol into RA. DHRS9 expression was also increased following induction of viral infection in EBV-positive Burkett lymphoma cells. Jones et al. show that the EBV early-intermediate protein BZLF1 activates the DHRS9 promoter through a direct DNA binding mechanism. BZLF1 expression in gastric carcinoma cells was also sufficient to activate DHRS9 gene expression and increased the ability of retinol to induce the RA-responsive gene CYP26A1. The authors suggest that production of RA during EBV infection may enhance viral replication by promoting keratinocyte differentiation.
11. Retinoids Influence the Factors That Both Inhibit and Contribute to Influenza Pathogenesis
Manicassamy and Pulandran [67] reviewed recent work on the role of retinoic acid in the regulation of immune responses by dendritic cells and macrophages. The dendritic cell plays a central role in the innate immune response, which occurs early after exposure to infectious agents, including influenza virus infection. Dendritic cells (DCs), derived from bone marrow, are found in the subepithelial layer of the respiratory tract and throughout the body. Their role is to identify and capture invading pathogens (bacteria, viruses, parasites, and fungi) through pattern (or pathogen) recognition receptors (PPRs), based on pathogen-associated molecular patterns. The several classes of antiviral PPRs include the Toll-like receptors (TLRs) and the retinoic acid-inducible gene-I-like receptors (RIG-I) [136]. The 13 TLRs [137] collectively sense a wide array of microbial stimuli, including influenza virus [138]. Intracellular TLR signaling within dendritic cells is mediated by several adapter proteins, including MyD88 and toll-interleukin-1-receptor domain-containing adapter-inducing interferon beta (TRIF). After decoding and integrating the signals generated by sensing microbial molecules within TLRs, the adapter proteins convey this information to naive antigen-specific T cells and B cells, thereby launching the adaptive immune response to invading pathogens [139].
Antiviral cytokine gene expression in epithelial cells induced by influenza A is triggered by RIG-I and mda-5, whose expression is positively regulated by IFN-alpha [140]. RIG-I and melanoma differentiation-associated gene (mda-5) function as receptors for double-stranded RNA. Both interferon (IFN)-alpha and IFN-beta strongly enhance RIG-I and mda-5 mRNA in DCs and epithelia. influenza A virus-induced RIG-I detects influenza after fusion and replication in infected cells [141]. Influenza genomic RNA is also recognized by Toll-like receptor (TLR) 7 [138, 142] although most DCs use the RIG-I pathway in response to virus infection [141]. Signaling through both RIG-I and TLR7 results in the production of IFNs, which limit viral replication and increase resistance to infection.
Influenza virus infection is associated with IL-1/? production in bronchoalveolar lavage luid of mice [143] and activates IL-1 /? and IL-18 production in human macrophages [144]. IL-1 is responsible for acute lung immunopathology but promotes survival of the mice after influenza virus infection [145].
DCs suppress immune responses through the generation of Foxp3+ regulatory T cells (Tregs) and fine-tune the response by altering the T-helper (Th)1/2/17 balance. Foxp3 is a transcription factor essential for the differentiation and function of Tregs [146]. TLRs are also expressed by Tregs, which inhibit TLR hyperactivity and prevent sepsis and autoimmune diseases, but direct TLR activation on Tregs can block Treg function and amplify immune responses [147]. Recent evidence suggests that the catalysis of vitamin A into retinoic acid (RA) in subsets of DCs is vital for the induction of Foxp3+ Tregs. RA generation in DCs also enhances IgA secretion by B cells ([67]; see Figure 2).
DCs in the lamina propria of the small intestine induce Tregs via a mechanism that depends on retinoic acid [148]. Conversion ofnaive T cells to T regulatory cells was impaired by adding inhibitors of retinal dehydrogenases, indicating that the RA produced by the DCs facilitated the conversion. However, RA alone failed to induce the conversion of naive T cells to T regulatory cells but did so in the presence of TGF-|3. In the mesenteric lymph node the DCs express aldh1a2, a retinal dehydrogenase involved in the conversion of retinol to retinoic acid [149], and induce T regulatory cells in the presence of TGF-/?. Certain subsets of DCs in the lamina propria of the small intestine induce robust T17 responses and promote the differentiation of Th17 cells.
Retinoic acid has a concentration-dependent effect in promoting Th17 responses: low doses (1 nM) stimulate Th17 responses whereas higher doses (10 nM) suppress both T17 and Th1 responses [150, 151]. Th17 cells synthesize and secrete IL-17, and a reciprocal relationship exists between Th17 cells and Tregs whereby the differentiation of induced Tregs and Th17 cells depends on retinoic acid via TGF-/5 dependent induction of Foxp3 [152]. It is known that DCs in the gut express aldh1a1 and aldh1a2 [153]. In contrast to the gut, although vitamin A is stored in the liver, lungs, and bone marrow [72], it is not known if DCs in these organs constitutively express vitamin A metabolizing enzymes in different subpopulations of APCs during influenza infection or other inflammatory disease conditions [67].
A study of cytokine proiles in patients with mild and severe new variant (nv) influenza A H1N1 infection during the irst ive days following infection [154] found high levels of type-II interferon (IFN-y) and of mediators of T-helper 17 (IL-8, IL-9, IL-17, IL-6) and T-helper 1 (TNF-a, IL-15, IL-12p70) responses exclusively among the hospitalized patients (n = 20). The hallmarks of critical illness were IL-15, IL-12p70, and IL-6, and they were inversely associated with a reduced partial pressure of oxygen (PaO2) in arterial blood. On the other hand, in the one patient who died (on day 5 after disease onset), there was a high viral load but undetectable serum levels of IL-17. T1 adaptive immunity is an important response against intracellular microbes such as viruses, and Th17 cells participate in host defense reactions as well as in tissue inflammation in several autoimmune diseases, allergic diseases, and asthma [155]. It remains unclear whether the indings indicate a detrimental or beneficial role for these T1- and T17-driven cytokine proiles in severe influenza. Th17 cells are associated with tissue inflammation in several autoimmune and allergic diseases, and vitamin D reduces the Vitamin A (retinol)
Figure 2: Retinoic acid synthesis pathway in dendritic cells and its effects on lymphocytes. Retinoic acid is produced from vitamin A (retinol) via a two-step enzymatic pathway that oxidizes retinol to retinaldehyde and then retinaldehyde to RA. Oxidation of retinol to retinaldehyde requires the activities of several alcohol dehydrogenises (ADH-1, -4,-5) and subsequently, retinaldehyde is oxidized to retinoic acid by retinal dehydrogenases (RALDH). RA produced by DCs acts on T and B lymphocytes and induces the mucosal homing receptors a4/37-integrin, and CCR9. RA in the presence of TGF-/3 promotes the conversion of naive T cells into Foxp3+ regulatory T cells and at high concentration inhibits the differentiation of Th-17 cells. In addition, RA synergizes with IL-6 and IL-5 and promotes class-switching to IgA in B cells (source: [67]).
number of IL-17 secreting cells [156]. Given the balance-type or inverse association between vitamins D and A, vitamin A could contribute to IL-17-secretion since the main target cells of IL-17 are neutrophils, and the latter are influenced by retinoids.
Neutrophils are the most prevalent white blood cells in the circulation and represent the first line of defense against invading microorganisms. Influenza virus induces inflammatory cytokines from primed human neutrophils, a process that requires endosomal acidification and viral uncoating; moreover, TLR-7 is essential for influenza recognition and inflammatory cytokine production by murine neutrophils and thus is critically involved in influenza-induced neutrophil activation [157]. In severe cases of influenza infection a pronounced neutrophil infiltrate is seen in the lungs, accompanied by intense cytokine and chemokine production [158]. Pluripotent stem cells in bone marrow differentiate into mature neutrophils, enter the blood stream, and die within 24 hr via apoptosis. Retinoids (retinoic acid, in particular) play a critical role in the differentiation and maturation of neutrophils [159]. RA directly modulates gene expression via binding to its nuclear receptors, which in turn activate transcription of genes that are essential for differentiation of immature cells to neutrophils. Involvement of RA receptors in the pathogenesis of acute promyelocytic leukemia (APL) reflects the important role played by these receptors in the differentiation of immature myeloid cells into neutrophils. APL is characterized by the t(15;17) chromosomal translocation and formation of the abnormal PML-RARa fusion protein [160]. Human neutrophils also contain defensins, a family of small cysteine-rich endogenous antimicrobial peptides that can inactivate influenza A virus [161]. The induction of defensins in epithelial cells is mediated by cell-surface TLRs [162].
Cathepsin proteases are primary neutrophil granule components and mediators of osteoclastic bone resorption and are potent catabolic agents that result in destruction of collagen, elastin, gelatin, and bone. Retinoic acid strongly upregulates neutrophil cathepsins. Superoxide is also induced by retinoids in neutrophils, contributing to the acute ability of the neutrophil to cause tissue damage. All-trans-retinoic acid (ATRA, <200 M) dose-dependently induces superoxide generation in neutrophils, contributing to the "retinoic acid syndrome" which occurs in up to 26% of ATRA-treated patients with acute promyelocytic leukemia. ATRA induces superoxide through both direct (transcriptional) and indirect mechanisms [163]. Conversely, neutrophil function is reduced in vitamin A-deficient animals due to a reduction of superoxide production, associated with increased infectivity [164]. Chemotherapy with ATRA is associated with hematologic, inflammatory, and immunologic reactions, including a Sweet's syndrome-like neutrophilic panniculitis (inflammation of subcutaneous adipose tissue) that occurs concomitantly with neutrophilic differentiation [165]. The mechanism of systemic spread of H5N1 virus in patients with avian influenza is unknown. Based on immunohistochemical findings of H5N1 nucleoprotein and hemagglutinin in the nucleus and cytoplasm of neutrophils in the placental blood of a pregnant woman, it was suggested that neutrophils could serve as a vehicle for viral replication and transportation in avian influenza [166].
12. Retinoic Acid Influences Factors Associated with Severe Influenza Infection
The foregoing review indicates that retinoids are importantly involved in many of the factors and mechanisms associated with the response to influenza viral infection. Could the severity of influenza be influenced in a concentration-dependent fashion by retinoic acid? It has been noted that low concentrations of retinoic acid stimulate and promote Th17 responses, whereas higher concentrations suppress both Th17 and Th1 responses [150]. Consistent with this observation, Bermejo-Martin et al. [154] reported low levels of IL-17 in hospitalized patients with severe influenza, suggesting high concentrations of circulating retinoids. In the same study, one of the hallmarks of critical illness was elevated IL-6, a finding reported earlier by Kaiser et al. [167]. Heltzer et al. [168] studied cytokine levels in 10 children with severe influenza and 24 noninfected controls and similarly found significantly increased plasma levels of IL-6 as well as IL-12 and IFN-y in patients versus controls. Based on parallel in vitro infection studies, the ability to produce TNF-a was also found to be compromised. It has been reported that a subset of dendritic cells in the lung has an intrinsic capacity to promote Th2 responses via their production of IL-6 [169]. Given that retinoic acid, via RAR, directly suppresses Th1 development and directly enhances T2 development [170], this observation provides further evidence linking retinoic acid and IL-6 production.
Vitamin A is also required for B-cell-mediated immu-noglobulin A (IgA) responses to bacterial polysaccaride antigens in addition to its role in the induction of T cells in the gut. Normal intestinal mucosa contains abundant IgA-secreting cells, which are generated from B cells in gut-associated lymphoid tissues (GALTs). DCs from GALT induce T-cell-independent expression of IgA and gut-homing receptors on B cells. GALT-DC-derived retinoic acid alone conferred gut tropism but could not promote IgA secretion. However, RA potently synergized with GALT-DC-derived IL-6 or IL-5 to induce IgA secretion [171]. IL-6 is also induced by IL-17 and contributes to the generation of Th17 cells [151]. RA and the IL-6 family of cytokines have a synergistic effect on astrogliogenesis in mouse neural precursor cells [172]. Glial fibrillary acidic protein (GFAP) is the main astroglial marker during astrogliogenesis. The canonical pathway for GFAP expression in astrocytes is triggered by binding of cytokines from the IL-6 family to their receptors, which subsequently activate the JAK/STAT intracellular pathway, leading to the expression of GFAP in astrocytes. Herrera et al. [173] reported that RARa links retinoic acid signaling to the cytokine-stimulated pathway leading to GFAP expression and also plays a key part in the synergistic actions of retinoic acid and IL-6 in this pathway. The specific RARa antagonist Ro 41-5253 also reduced cytokine-induced GFAP [173].
Studies of lung tissues from fatal infantile cases of influenza provide evidence of robust viral replication and heightened expression of markers of apoptosis (programmed cell death). High titers of influenza induce DC apoptosis [174]. Severe infantile RSV and influenza virus infection of the lower tract respiratory tract is characterized by inadequate adaptive immune response, robust viral replication, and apoptotic crisis [175]. Infection of human airway epithelial cells with human rhinovirus, both in vitro and in vivo, increases the expression of inducible nitric oxide (NO) synthase, which correlates with increased levels of NO in exhaled air. Te common cold is triggered by a variety of viral pathogens, mostly rhinoviruses; complications include sinusitis, otitis media, and exacerbations of asthma and chronic obstructive lung disease. NO inhibits human rhinovirus-induced epithelial expression of several proin-flammatory cytokines; it inhibits viral replication in epithelial cells in vitro and modulates several signal transduction pathways associated with cytokine generation; it is also involved in the nitrosylation of viral proteases and interacts with the immune system. Increased NO generation during rhinovirus infections is associated with fewer symptoms, more rapid improvement in symptoms, and more rapid viral clearance [176]. In experimental studies in A/NWS/33 influenza-infected BALB/c mice, marked increases were reported in the levels of metabolites of NO in the olfactory bulb and hippocampus. influenza infection was associated with the activation of astrocytes, the major cell type in the brain, as well as NO synthase (NOS-2) expression, which in turn enhanced NO production and the expansion of capillary blood vessels [177].
Observations on the roles of apoptosis and nitric oxide (NO) in colds and influenza are relevant to the hypothesis that the severity of influenza infection is associated with increased retinoic acid concentrations. Retinoids are inversely related to and suppress cytokine-induced production of NO, which is antiapoptotic, whereas retinoic acid is proapoptotic [178]. Nitric oxide is a potent vasodilator, generated enzymatically from L-arginine by the action of nitric oxide synthase (NOS) [179]. All-trans-RA blocks the release of tumor necrosis factor from peritoneal macrophages stimulated with endo-toxin and interferon-gamma and inhibits NO production in these cells [180]. All-trans-RA and its active analogues also block cytokine-stimulated expression of inducible NO in cultured vascular smooth muscle cells and isolated rat aortic rings [181]. 9-cis RA, an agonist of both RARs and RXRs, suppressed the production of TNF-alpha and NO in a concentration-dependent manner in lipopolysaccharide-stimulated rat Kupffer cells in vitro [182]. RA also regulates NO production in rat aortic smooth muscle cells, and the latter expresses NOS-2 in response to proinflammatory cytokines. RA inhibited IL-1-beta-induced NOS-2 mRNA expression and NO production. These effects were attenuated by the retinoic acid receptor (RAR) antagonist CD3106, indicating that they were mediated through the RARs [183]. Thus, NO may be therapeutically effective in colds (and possibly influenza) by inhibiting the actions of retinoids.
13. Role of the Liver in Severe Influenza A Infection
A clue to understanding severe or lethal influenza may lie in observations implicating the liver, the main storage organ for vitamin A, in influenza infection. In addition to being the target of viruses that are specifically hepatotropic, for example, hepatitis A, B, C, and E, the liver can also be affected as part of a generalized host infection with viruses, including influenza, which primarily target other tissues, notably the upper respiratory tract [112]. Mild infection is thought to cause few extrapulmonary manifestations. However, gastrointestinal complications in more severe cases of influenza A can include pancreatitis, hemorrhagic gastritis, Reye's syndrome, and acute hepatic decompensation in adults with underlying chronic liver disease [184]. Cases have been reported of acute hepatitis with or without liver failure in previously healthy children with influenza A infection, none of whom had received aspirin or other known hepatotoxic drugs except acetaminophen. This led Whitworth et al. [113] to suggest that influenza A may be hepatotropic and that children with severe influenza A should be screened for liver injury.
Respiratory infections in humans, including influenza, are often accompanied by mild hepatitis, although the mechanisms are not well understood. In their study of patients who were hospitalized with severe nvH1N1 infection, Bermejo-Martin et al. [154] found that, in addition to the symptoms of fever, cough, headache, tiredness, myalgia, and dyspnea, lactate dehydrogenase and transaminases were raised fourfold above those of hospitalized but noncritical illness controls, providing evidence of necrotic damage in the liver. Polakos et al. [111] infected 15 subjects intranasally with influenza A (H1N1), and 4/15 developed elevated serum transaminases (>3 times the upper limit in 2 subjects), suggesting clinically significant hepatitis. Te rise in liver enzymes occurred after the decline in fever, implying that liver involvement was not caused by the initial viral replication and activation of the innate immune system. Hepato-cytes were found to be damaged by viral specific CD8+ T cells generated in the lungs, a condition Polakos et al. described as "collateral damage", since there was no evidence of influenza viral antigen in the liver.
Te model proposed here aims to explain the mechanism of influenza A-associated liver dysfunction and its role in increasing the severity of infection. It is consistent with the overall low vitamin D:A ratio hypothesis of severe influenza and involves alterations in retinoid metabolism. It is suggested that influenza-induced liver involvement worsens the outcome of infection via the hepatic release of unbound retinyl esters and retinoic acids which are transported to and damage the lung as well as other organs, there by contributing to the development of pneumonia, heart and kidney failure, and sepsis.
13.1. Outline of the Model. (1) Viral-specific CD8+ T cells generated in response to influenza infection outside the liver trigger cell-mediated apoptotic hepatitis [111]. Tis process may occur through infection-induced activation of the retinoid cascade in the liver, leading to increased retinoic acid production and RAR activation and expression within the hepatic cell nuclei.
In their study of experimentally induced influenza, Polakos et al. [111] found evidence of clinically significant hepatitis and hypothesized that the latter was due to the formation of inflammatory foci that included apoptotic hepatocyte antigen specific CD8+ T cells and Kupffer cells. Using a mouse model of influenza, Polakos et al. found that the severity of hepatitis correlated with the magnitude of the antiviral CD8+ T cells response despite the lack of detectable virus in the liver; they also noted that the hepatitis was markedly less severe in the absence of Kupffer cells, which they attributed to the loss of resident macrophages.
According to the model proposed here, influenza-induced increased RAR activation inhibits the production and secretion of retinol binding-protein (RBP) via a process of feedback inhibition [185], thereby lowering serum retinol concentrations and simultaneously increasing the accumulation and expression of retinoids in the liver. Tese changes induce inflammation and tissue damage, consistent with the known role of excessive vitamin A in inducing liver damage [186] and apoptosis [187]. The suggested mechanism oftissue damage involves Kupffer cells which, in addition to being the source of macrophages in the liver, also play a critical role in hypervitaminosis A. Vitamin A activates Kupffer cells through INF-y production by activated T lymphocytes [188], and activated Kupffer cells potentiate liver toxicity even with low levels of hepatotoxins such as alcohol [189]. Hence, the observation of Polakos et al. [111] that the severity of hepatitis in mice infected with influenza was reduced in the absence of Kupffer cells could be due to the fact that these cells play a critical role in the processes mediating vitamin A intoxication.
Vitamin A hepatotoxicity is associated histologically with the accumulation of perisinusoidal lipocytes, associated i-brosis, obstruction of sinusoids and terminal venules, sclerosis of central veins, atrophy of adjacent hepatocytes, and an increase in basement membrane-like material and collagen within the perisinusoidal space in association with lipid-filled Ito cells [190-192]. The nonparenchymal Ito (or stellate) cells in the liver contain about 90% of the total vitamin A reserves of the body and appear to be the primary source of the extracellular matrix deposition in liver ibrosis and of the apoptotic mediators involved in fibrosis [193]. During hepatic fibrosis, the normally quiescent vitamin A-storing stellate cells transform into myoibroblastic cells and lose their intracellular droplets of retinyl esters, the storage form of vitamin A. Te loss of retinyl esters is associated with increased RA formation, which in turn facilitates TGF-beta-mediated liver fibrogenesis [194]. This phenotypic transformation induces potent proinflammatory and profibrogenic activities and is underpinned by changes in the expression of numerous genes [195].
(2) Retinoid induced Kupffer cell activation and hepa-totoxicity, lead to a transient form of cholestatic liver dysfunction, a condition in which bile regurgitates into the circulation, raising the level of all biliary substances in the blood [192,196].
In cholestasis, vitamin A metabolites and bile acids are refluxed from the liver and stored retinyl esters leak into the circulation from damaged hepatocytes. Metabolites of retinol, retinyl acetate, retinal, retinoic acid, retinol beta-glucuronide, retinotaurine, and other unidentified vitamin A compounds are excreted in the bile [79, 197] in proportion to the total liver stores of the vitamin [198]. A variety of infections and stressors are associated with transient declines in plasma retinol concentrations [199, 200], but plasma retinol concentrations tend to rebound when the inflammatory stimulus is removed [201].
The hypothesized net effect of transient cholestasis in influenza A infection is high circulating concentrations of retinoid metabolites and retinyl esters, with low or normal concentrations of retinol and RBP. In studies of infective hepatitis, the RBP-prealbumin complex and plasma vitamin A were decreased in association with reduced hepatic mobilization and very high liver levels of vitamin A, resulting in a mixed symptom pattern of hypo- and hypervitaminosis A [202].
(3) Retinoic acid and stored retinyl esters released into the circulation from damaged liver cells cause acute lung injury (ALI) via apoptosis and necrosis of lung tissue.
The symptoms of ALI involve the destruction of alveolar epithelial tissue, diffuse alveolar damage with hyaline membrane formation, bronchiolitis with squamous metaplasia, and pulmonary congestion. At the molecular level the patho-genesis of ALI includes acute neutrophilic infiltration and proinflammatory cytokine secretion in the lungs [5]. Cell death and injury due to apoptosis and necrosis are seen in the lung during the pathogenesis of ALI/ARDs. Apoptosis in the alveolar epithelial cells of influenza H5N1 virus-infected humans suggests that it plays a major role in influenza pathogenesis in humans by destroying alveolar epithelial cells [203]. Apoptosis of lymphocytes has been reported in mice infected with lethal influenza H5N1 virus [204], but the mechanism is unclear. Neutrophil counts may be high in the initial phases of influenza infection but are often very low in bacterial pneumonia that can occur as a sequel to influenza infection [16]. While increased retinoid may drive the early increase in neutrophil count, a point may be reached where rising concentrations of circulating retinoid compounds as a result of liver damage could induce massive apoptosis of the neutrophils and lymphocytes. The net effect would be to lower the functionality of the immune system, leading to progressive bacterial infection and sepsis.
The three recognized stages of sepsis, severe sepsis, and septic shock are proinflammatory and procoagulant responses to invading pathogens associated with a progressively increased risk of multiorgan failure and death. In cirrhosis, sepsis is accompanied by a markedly imbalanced cytokine response "cytokine storm" in which responses that are normally beneficial for fighting infections are converted into excessive, damaging inflammatory responses [205]. The molecular mechanisms of this excessive proinflammatory response are poorly understood. In patients with cirrhosis and severe sepsis, proinflammatory cytokines are thought to play a role in worsening liver function and the development of shock, renal failure, acute lung injury or acute respiratory distress syndrome, coagulopathy, hepatic encephalopathy, hyperglycemia, defective arginine-vasopressin secretion, and adrenal insufficiency [206].
The suggested association between liver damage-induced sepsis and alterations in retinoid metabolism is supported by a study of serum and hepatic vitamin A levels in patients with cirrhosis [207], in which serum retinyl ester concentrations were significantly higher than those in controls (Table 2). However, the percentage of serum retinyl esters as a fraction of total vitamin (retinol plus esters), which was not calculated but is an accepted indicator of vitamin A toxicity when >10%, was 20%, that is, double the percentage criterion for vitamin A toxicity.
A low vitamin D: A ratio has been postulated to increase the severity of influenza infection. Te risk of severe illness may be further exacerbated by the effect of illness on the liver and its stored contents of vitamin A. On this model, activation of the retinoid cascade causes liver damage and the spillage of retinoic acid and unbound retinyl esters into the circulation. It is hypothesized that this induces lung damage, multiorgan failure, and sepsis. Given the toxicity of retinoids and the fact that retinoid stores in the liver are sufficient to last the average adult for about two years [79], the presumably high quantity of retinoids spilled into the circulation in cholestatic conditions, including influenza, has the potential to cause considerable tissue damage.
(4) The mechanisms of tissue damage following the release of retinoids from the liver in influenza infection also could be due to the hepatic release of the enzyme xanthine oxidase (XO), which affects vitamin A metabolism.
Mice infected intranasally with influenza virus have high levels of XO, TNF, and IL-6 in bronchoalveolar lavage within 3 d after infection, as well as high levels of XO in serum and lung tissue [208, 209]. In humans, XO plays an important role in the catabolism of purines and is normally found in the liver but is released into the blood during severe liver damage [210]. XO catalyzes the oxidation of hypoxanthine to xanthine and ofxanthine to uric acid. XO can also catalyze the conversion of retinaldehyde to retinoic acid [211]. Hence, increased circulating XO may be associated with increased concentrations of retinoic acid.
(5) Preexisting liver disease, conditions associated with liver dysfunction such as pregnancy, and metabolic conditions including obesity and diabetes are strongly associated with an increased risk of severe influenza A infection [212214]. It is proposed that susceptibility to severe infection induced by these conditions is due to liver dysfunction and associated alterations in retinoid metabolism.
Table 2: Serum vitamin A concentrations in controls and in patients with cirrhosis.
RBP: retinol-binding protein (source: Ukleja et al. [207]).
In a study of 63 patients hospitalized with the 2009 H1N1 influenza infection at the Mayo Clinic in Rochester, Minnesota, the three most common comorbidities were hypertension (47%), obesity (44%), and diabetes mellitus (32%) [215]. The present hypothesis—that alterations in retinoid metabolism in these conditions contribute to increased risks of severe influenza infection—is consistent with growing evidence of a role for retinol-binding protein in diabetes [216-219]. Retinoid profiles were studied in a rat model of gestational diabetes, preeclampsia and fetal growth restriction by the present author and colleagues [220]. Liver damage, indicated by elevated liver enzymes, was accompanied by significantly lower median plasma retinol (ROL) concentration but also by a significantly higher retinyl ester-to-ROL ratio, a significantly higher percent retinyl ester level (median, 24% versus 11%; P = 0.008), and a significantly higher concentration of all-trans-RA in the experimental rats compared to the controls. These results suggest that gestational diabetes, preeclampsia and fetal growth restriction may be associated with an endogenous form of retinoid toxicity associated with impaired liver function.
14. Implications for Prevention and Treatment
Efforts to prevent and treat influenza have focused primarily on the virus [221]. Since antiviral agents are expensive and in short supply, identifying effective, inexpensive and universally available antiviral agents should be a high priority [222]. However, the development of vaccines and antiviral compounds is complicated by frequent mutations and gene reassortments between viruses, suggesting that approaches aimed at the host (e.g., developing cost-effective preventive or therapeutic agents) may hold greater promise [223]. In fact, it is increasingly accepted that illness and pathology in infectious diseases in general are due less to the invading organism than to the body's response to infection [205].
Evidence presented in this paper suggests that both vitamins A and D are importantly involved in the host defense mechanisms that regulate the severity of influenza and other viral illnesses. The presence of high preexisting retinoid concentrations associated with increased tissue stores and/or dietary intake (i.e., a low vitamin D: A ratio) may increase host susceptibility and disease severity via the destruction of mucosal barriers and the resulting bacterial invasion and sepsis—processes that appear to be worsened by liver damage and comorbidities affecting the liver. Conversely, reduced but not necessarily deficient background retinoid concentrations (i.e., a high vitamin D: A ratio) could have the opposite effect of reducing disease susceptibility, lowering disease severity, and improving disease outcomes. These hypotheses have implications for the prevention and treatment of influenza virus infections.
The present review suggests that in addition to vitamin D supplementation, based on the vitamin D deficiency hypothesis of influenza [33-35], measures targeting retinoids to reverse the low vitamin D : A ratio may also be useful in prevention and treatment. Such measures could include, for example, modest exposure to solar radiation, to increase vitamin D and lower vitamin A, dietary changes to lower the intake and accumulation of vitamin A, and the use of pharmacological agents to affect both vitamin A and D metabolism.
With regard to dietary measures, the literature suggests that influenza-induced or preexisting liver disease may significantly worsen the outcome of influenza infection.
The increased risk of severe influenza on the part of individuals with diabetes and the obese may be related to liver dysfunction and alterations in retinoid metabolism, as suggested above. Aging is also associated with an increased risk of hospitalization and death from influenza-associated pneumonia, and people of age 65 and older account for >90% of influenza-related deaths [224]. Could aging be associated with increased susceptibility to severe influenza due to a progressive accumulation of retinoids with advancing age? Several observations support this hypothesis: serum levels of vitamin A increase in elderly populations [225, 226]; intracellular retinoids and retinoid-binding proteins are present in leucocytes (granulocytes and mononuclear cells) and increase quadratically with age; serum concentrations of retinoic acid increase in the elderly [227]; vitamin A absorption is also increased in elderly humans and animals [228, 229].
If this hypothesis is correct, dietary restriction could influence the occurrence and severity of influenza. Indeed, it is known that restricting caloric intake to 60%-70% of normal adult weight prolongs lifespan 30%-50% and confers excellent health across a broad range of species; every other day feeding produces similar effects in rodents. Based on such observations and on self-experimentation, Johnson et al. [230] proposed that alternate-day calorie restriction (one day consuming 20%-50% of estimated daily caloric requirements, the next day eating ad libitum) would result in health beneits starting in as little as two weeks, with declines in insulin resistance, asthma, seasonal allergies, infectious diseases of viral, bacterial, and fungal origin, and many other conditions. They suggest that widespread use of this pattern of eating could affect epidemics of influenza and other communicable diseases by improving resistance to infection.
With regard to pharmacological measures , xanthine oxidase (XO) inhibitors are reported to have a potent antiviral effect against influenza A. A mouse model of influenza was associated with a 400-fold increase in XO activity, measured per volume of alveolar lavage luid, as well as increased uric acid levels in serum. Treatment with allopurinol (an inhibitor of XO) and chemically modified superoxide dismutase (a scavenger of O2-) improved the survival rate of influenza virus-infected mice. These results were thought to indicate that oxygen-free radicals produced by XO contributed to the pathogenesis of influenza virus infection in mice and that antioxidant therapy could eliminate oxygen radicals [231]. As noted, XO converts hypoxanthine to xanthine and xanthine to uric acid and also catalyzes the conversion of retinaldehyde to retinoic acid. Allopurinol also has a dose-dependent inhibitory effect on the synthesis of retinoic acid from xanthine [211]. The efficacy of allopurinol as a treatment for influenza could be due in part to the effect of XO on the metabolism of retinoids.
Bassaganya-Riera et al. [223] suggested using peroxisome proliferator-activated receptor (PPAR) agonists to down-regulate the inflammatory response to respiratory viruses. Noting that excessive inflammatory responses to respiratory viruses "cytokine storms" are due to immune dysregulation and that PPARs play important roles in antagonizing core inflammatory pathways such as NF-jcB, AP1, and STAT, the authors proposed that PPARy agonists could be used to downregulate inflammatory responses to respiratory virus-related pulmonary inflammation. It is of interest in this connection, given the proposed role of retinoic acid in influenza, that the first RARa antagonist (Ro41-5253) to be developed [232] is a PPARy agonist insofar as it binds to and activates PPARy [74]. This suggests that expression of PPAR]' is effectively equivalent to RARa antagonism; that is, RARa antagonism is a natural consequence of PPARy activation or expression.
15. Conclusion
The severity of influenza virus infection can perhaps be considered a function of two sets of opposing factors. On one side are the factors that fight infection, exerting inhibitory effects on the virus at different stages of the disease process. On the other side are the factors that drive the disease, allowing the virus to multiply, spread, and induce symptoms. There is evidence that retinoids play a central role in both processes. It has been proposed that (1) reduced exposure to sunlight and/or preexisting vitamin D deficiency simultaneously increase the accumulation and potential toxicity of endogenous retinoids, while the decreased vitamin D-to-vitamin A ratio triggers viral activation or increases susceptibility to novel strains of influenza virus; (2) increased but normal physiological concentrations of retinoid effectively work with vitamin D to inhibit influenza pathogenesis; and (3) higher background concentrations of vitamin A (i.e., very low vitamin D: A ratios), for example, associated with diseases and conditions affecting the liver, enhance viral replication and increase the likelihood of severe or lethal complications of the disease. It is possible and indeed likely that, on the vitamin A side, different retinoid receptor isotypes (RARs, RXRs) will prove to be involved in the outcomes of altered vitamin D: A ratios, either enhancing or inhibiting infection and viral replication, depending on the background level of retinoids in the body. For instance, one group of retinoid receptor isotypes may facilitate and exacerbate influenza viral infections, while another set may come into play to inhibit viral proliferation and infection.
In summary, it is proposed that lack of solar radiation and/or vitamin D deficiency increase the availability and potential toxicity of retinoids, and the latter interact with and induce viral activation at the genome level to trigger influenza. On this hypothesis, influenza viral pathogenesis involves both vitamin D deficiency and endogenous retinoid overexpression. In seasonal influenza, ever-present influenza viruses may be activated and disease symptoms triggered by declining vitamin D concentrations and worsened by retinoid accumulation and overexpression. In pandemic influenza, while the virulence of the strain of virus may account for disease epidemicity, the likelihood of particular individuals being infected may depend on the background nutritional status of vitamin A and D, whereby infectivity may be reduced by vitamin D supplementation but enhanced by vitamin A supplementation or excess. Retinoid receptor overexpression may thus contribute to the pathogenesis of influenza and related viral infections, causing an endogenous form of hypervitaminosis A that manifests itself in the symptoms of the disease.
The model could be tested by determining the presence and concentration of retinoids in the secretions of patients with influenza, particularly retinyl esters and retinoic acid. Alterations in serum retinoid profiles could also be examined in case-control studies. Subject to obtaining support for the model, clinical trials could be initiated to determine the relative therapeutic efficacy of vitamin D supplementation, retinoic acid receptor antagonists, and the combination of vitamin D supplementation plus a retinoic acid receptor antagonist. The formal study of retinoid profiles and expression in influenza infection is, however, a new field awaiting exploration.
Acknowledgments
The author thanks Hilary Butler and Ronald Eccles for comments on earlier versions of this paper, and Xueyuan Wang, MPH, for assistance in preparing the paper for publication.
References
[1] R. G. Webster, W. J. Bean, O. T. Gorman, T. M. Chambers, and Y. Kawaoka, "Evolution and ecology of influenza A viruses," Microbiological Reviews, vol. 56, no. 1,pp. 152-179, 1992.
[2] A. D. Langmuir and S. C. Schoenbaum, "The epidemiology of influenza," Hospital Practice, vol. 11, no. 10, pp. 49-56, 1976.
[3] L. Simonsen, "The global impact of influenza on morbidity and mortality," Vaccine, vol. 17, supplement 1, pp. S3-S10, 1999.
[4] W. W. Thompson, D. K. Shay, E. Weintraub et al., "Mortality associated with influenza and respiratory syncytial virus in the United States," Journal of the American Medical Association, vol. 289, no. 2, pp. 179-186, 2003.
[5] H. L. Wang and C. Y. Jiang, "Avian influenza H5N1: an update on molecular pathogenesis," Science in China, vol. 52, no. 5, pp. 459-463, 2009.
[6] P. LaRussa, "Pandemic novel 2009 H1N1 influenza: what have we learned?" Seminars in Respiratory and Critical Care Medicine, vol. 32, no. 4, pp. 393-399, 2011.
[7] E. Spackman, "A brief introduction to the avian influenza virus," Methods in Molecular Biology, vol. 436, pp. 1-6, 2008.
[8] D. B. Lewis, "Avian flu to human influenza" Annual Review of Medicine, vol. 57, pp. 139-154, 2006.
[9] G. M. Air and W. G. Laver, "The neuraminidase of influenza virus," Proteins, vol. 6, no. 4, pp. 341-356, 1989.
[10] M. Schoch-Spana, "Implications of pandemic influenza for bioterrorism response," Clinical Infectious Diseases, vol. 31, no.6, pp. 1409-1413,2000.
[11] A. W. Mounts, H. Kwong, H. S. Izurieta et al., "Case-control study of risk factors for avian influenza A (H5N1) disease, Hong Kong, 1997," Journal of Infectious Diseases, vol. 180, no. 2, pp. 505-508, 1999.
[12] G. Neumann, T. Noda, and Y. Kawaoka, "Emergence and pandemic potential of swine-origin H1N1 influenza virus," Nature, vol. 459, no. 7249, pp. 931-939, 2009.
[13] D. M. Morens, J. K. Taubenberger, and A. S. Fauci, "The 2009 H1N1 pandemic influenza virus: what next?" MBio, vol. 1, no.
4, article e00211-10, 2010. *[14] R. Eccles, "Understanding the symptoms of the common cold and influenza," Lancet Infectious Diseases, vol. 5, no. 11, pp. 718-725,2005.
[15] D. S. C. Hui, "Review of clinical symptoms and spectrum in humans with influenza A/H5N1 infection," Respirology, vol. 13, supplement 1, pp. S10-S13, 2008.
[16] T. M. Tumpey and J. A. Belser, "Resurrected pandemic influenza viruses," Annual Review of Microbiology, vol. 63, pp. 79-98, 2009.
[17] D. M. Morens, J. K. Taubenberger, and A. S. Fauci, "Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness," Journal of Infectious Diseases, vol. 198, no. 7, pp. 962-970, 2008.
[18] A. N. Abdel-Ghafar, T. Chotpitayasunondh, Z. Gao et al., "Update on avian influenza A (H5N1) virus infection in humans," New England Journal of Medicine, vol. 358, no. 3, pp. 261-273,2008.
[19] J. Gu, Z. Xie, Z. Gao et al., "H5N1 infection of the respiratory tract and beyond: a molecular pathology study," The Lancet, vol. 370, no. 9593, pp. 1137-1145, 2007.
[20] M. D. de Jong, "H5N1 transmission and disease: observations from the frontlines," The Pediatric Infectious Disease Journal, vol. 27, no. 10, pp. S54-S56, 2008.
[21] N. L. la Gruta, K. Kedzierska, J. Stambas, and P. C. Doherty, "A question of self-preservation: immunopathology in influenza virus infection," Immunology and Cell Biology, vol. 85, no. 2, pp. 85-92, 2007.
[22] T. Henry, A. Brotcke, D. S. Weiss, L. J. Tompson, and D. M. Monack, "Type I interferon signaling is required for activation of the inflammasome during Francisella infection," Journal of Experimental Medicine, vol. 204, no. 5, pp. 987-994, 2007.
[23] T. T. Wang and P. Palese, "Unraveling the mystery of swine influenza virus," Cell, vol. 137, no. 6, pp. 983-985, 2009.
[24] T. L. Brammer, E. L. Murray, K. Fukuda, H. E. Hall, A. Klimov, N. J. Cox et al., "Surveillance for influenza—United States, 1997-98, 1998-99, and 1999-00 seasons," Morbidity and Mortality Weekly Report, vol. 51, no. 7, pp. 1-10, 2002.
[25] D. M. Morens, "influenza-related mortality: considerations for practice and public health," Journal ofthe American Medical Association, vol. 289, no. 2, pp. 227-229, 2003.
[26] T. Jefferson, C. Di Pietrantonj, A. Rivetti, G. A. Bawazeer, L. A. Al-Ansary, and E. Ferroni, "Vaccines for preventing influenza in healthy adults," Cochrane Database ofSystematic Reviews, vol. 7, article CD001269, 2010, Update of: Cochrane Database of Systematic Reviews 2007; (2): CD001269.
[27] M. T. Osterholm, N. S. Kelley, A. Sommer, and E. A. Belongia, "Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis," The Lancet Infectious Diseases, vol. 12, no. 1,pp. 36-44, 2012.
[28] G. Dumyati and A. R. Falsey, "Antivirals for influenza: what is their role in the older patient?" Drugs and Aging, vol. 19, no. 10,pp. 777-786, 2002.
[29] A. Moscona, "Neuraminidase inhibitors for influenza," New England Journal of Medicine, vol. 353, no. 13, pp. 1363-1373,2005.
[30] M. D. de Jong, T. T. Tanh, T. H. Khanh et al., "Oseltamivir resistance during treatment of influenza A (H5N1) infection," New England Journal ofMedicine, vol. 353, no. 25, pp. 2667-2672, 2005.
[31] R. D. Smith, "Respondingto global infectious disease outbreaks: lessons from SARS on the role of risk perception, communication and management," Social Science and Medicine, vol. 63, no. 12, pp. 3113-3123,2006.
[32] J. S. M. Peiris, C. M. Chu, V. C. C. Cheng et al., "Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study," Te Lancet, vol. 361, no. 9371, pp. 1767-1772, 2003.
[33] J. J. Cannell, R. Vieth, J. C. Umhau et al., "Epidemic influenza and vitamin D," Epidemiology and Infection, vol. 134, no. 6, pp. 1129-1140,2006.
[34] J. J. Cannell, R. Vieth, W. Willett et al., "Cod liver oil, vitamin A toxicity, frequent respiratory infections, and the vitamin D deficiency epidemic," Annals of Otology, Rhinology and Laryngology, vol. 117, no. 11, pp. 864-870, 2008.
[35] J. J. Cannell, M. Zasloff, C. F. Garland, R. Scragg, and E. Gio-vanucci, "On the epidemiology of influenza," Virology Journal,vol. 5, article 29, 2008.
[36] J. J. Cannell, "Epidemic influenza and vitamin D," http:// www.medicalnewstoday. com / medicalnews. php ? newsd =51913. *[37] T. T. Wang, F. P. Nestel, V. Bourdeau et al., "Cutting edge: 1,25-Dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression," Journal of Immunology, vol. 173, no. 5 pp. 2909-2912,2004.
[38] R. E. Hope-Simpson and D. B. Golubev, "A new concept of the epidemic process of influenza A virus," Epidemiology and Infection, vol. 99, no. 1, pp. 5-54, 1987.
[39] R. E. Hope-Simpson, The Transmission of Epidemic Influenza, Plenum Press, New York, NY, USA, 1992.
[40] T. Jefferson, "Influenza vaccination: policy versus evidence" British Medical Journal, vol. 333, no. 7574, pp. 912-915, 2006.
[41] P. G. Szilagyi, G. Fairbrother, M. R. Griin et al., "influenza vaccine effectiveness among children 6 to 59 months of age during 2 influenza seasons: a case-cohort study," Archives of Pediatrics & Adolescent Medicine, vol. 162, pp. 943-951, 2008.
[42] N. J. Cox and K. Subbarao, "Influenza," The Lancet, vol. 354, no. 9186, pp. 1277-1282, 1999.
[43] E. Hypponen and C. Power, "Hypovitaminosis D in British adults at age 45 y: nationwide cohort study of dietary and lifestyle predictors," American Journal of Clinical Nutrition, vol. 85, no. 3, pp. 860-868, 2007.
[44] D. MacDonald and R. Swaminathan, "Seasonal variation in 25-OH vitamin D in plasma of Hong Kong Chinese," Clinical Chemistry, vol. 34, no. 11, p. 2375, 1988.
[45] A. W. C. Kung and K. K. Lee, "Knowledge of vitamin D and perceptions and attitudes toward sunlight among Chinese middle-aged and elderly women: a population survey in Hong Kong," BMC Public Health, vol. 6, article 226, 2006.
[46] H. I. Hall, D. S. May, R. A. Lew, H. K. Koh, and M. Nadel, "Sun protection behaviors of the U.S. white population," Preventive Medicine, vol. 26, no. 4, pp. 401-407, 1997.
[47] I. Laaksi, J. P. Ruohola, P. Tuohimaa et al., "An association of serum vitamin D concentrations < 40nmol/L with acute respiratory tract infection in young Finnish men," American Journal of Clinical Nutrition, vol. 86, no. 3, pp. 714-717, 2007.
[48] M. F. Holick, "Vitamin D deficiency," The New England Journal of Medicine, vol. 357, pp. 266-281, 2007.
[49] A. F. Gombart, N. Borregaard, and H. P. Koefler, "Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3," The FASEB Journal, vol. 19, no. 9, pp. 1067-1077, 2005.
[50] J. F. Aloia and M. Li-Ng, "Re: epidemic influenza and vitamin D," Epidemiology and Infection, vol. 135, no. 7, pp. 1095-1098, 2007.
[51] J. S. Adams and M. Hewison, "Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity," Nature Clinical Practice Endocrinology and Metabolism, vol. 4, no. 2, pp. 80-90, 2008.
[52] M. Zasloff "Antimicrobial peptides of multicellular organisms" Nature, vol. 415, no. 6870, pp. 389-395, 2002.
[53] E. Leikina, H. Delanoe-Ayari, K. Melikov et al., "Carbohydrate-binding molecules inhibit viral fusion at the airway surface by transactivation of the epithelial growth factor receptor" Nature Immunology, vol. 6, pp. 995-1001, 2005.
[54] D. M. Laube, S. Yim, L. K. Ryan, K. O. Kisich, and G. Diamond, "Antimicrobial peptides in the airway" Current Topics in Microbiology and Immunology, vol. 306, pp. 153-182, 2006.
[55] J. Schauber, R. A. Dorschner, A. B. Coda et al., "Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism," Journal of Clinical Investigation, vol. 117, no. 3, pp. 803-811, 2007.
[56] P. T. Liu, S. Stenger, H. Li et al., "Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response," Science, vol. 311, no. 5768, pp. 1770-1773, 2006.
[57] L. Sun, C. M. Finnegan, T. Kish-Catalone et al., "Human j3-defensins suppress human immunodeficiency virus infection: potential role in mucosal protection," Journal of Virology, vol. 79, no. 22, pp. 14318-14329, 2005.
[58] M. E. Klotman and T. L. Chang, "Defensins in innate antiviral immunity" Nature Reviews Immunology, vol. 6, no. 6, pp. 447-456, 2006.
[59] D. Bruce, J. H. Ooi, S. Yu, and M. T. Cantorna, "Vitamin D and host resistance to infection? Putting the cart in front of the horse" Experimental Biology and Medicine, vol. 235, no. 8, pp. 921-927, 2010.
[60] M. Urashima, T. Segawa, M. Okazaki, M. Kurihara, Y. Wada, and H. Ida, "Randomized trial of vitamin D supplementation to prevent seasonal influenza A in schoolchildren," American Journal of Clinical Nutrition, vol. 91, no. 5, pp. 1255-1260,2010.
[61] T. L. Clemens, J. S. Adams, S. L. Henderson, and M. F. Holick, "Increased skin pigment reduces the capacityofskin to synthesise vitamin D3," The Lancet, vol. 1, no. 8263, pp. 74-76, 1982.
[62] B. Srivastava, P. Blazejewska, M. Hessmann et al., "Host genetic background strongly influences the response to influenza a virus infections" PLoS One, vol. 4, no. 3, Article ID e4857,2009.
[63] C. Hoffmann and G. Eichele, "Retinoids in development" in The Retinoids: Biology, Chemistry, and Medicine, M. B. Sporn, A. B. Roberts, and D. S. Goodman, Eds., pp. 387-441, Raven Press, New York, NY, USA, 1994.
[64] M. Theodosiou, V. Laudet, and M. Schubert, "From carrot to clinic: an overview ofthe retinoic acid signaling pathway," Cellular and Molecular Life Sciences, vol. 67, no. 9, pp. 1423-1445, 2010.
[65] M. A. Lane and S. J. Bailey, "Role of retinoid signalling in the adult brain," Progress in Neurobiology, vol. 75, no. 4, pp. 275-293, 2005.
[66] G. Litwack, Ed., Vitamin A: Vitamins and Hormones, vol. 75, Elsevier Academic Press, San Diego, Calif, USA, 2007.
[67] S. Manicassamy and B. Pulandran, "Retinoic acid-dependent regulation of immune responses by dentritic cells and macrophage," Semin Immunol, vol. 21, pp. 22-27, 2009.
[68] D. J. Mangelsdorf, C. Thummel, M. Beato et al., "The nuclear receptor super-family: the second decade," Cell, vol. 83, no. 6, pp. 835-839, 1995.
[69] P. Huang, V. Chandra, and F. Rastinejad, "Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics," Annual Review ofPhysiology, vol. 72, pp. 247-272, 2010.
[70] M. Pfahl and F. Chytil, "Regulation of metabolism by retinoic acid and its nuclear receptors," Annual Review ofNutrition, vol. 16, pp. 257-283, 1996.
[71] C. B. Nilsson and H. Hakansson, "The retinoid signaling system—a target in dioxin toxicity," Critical Reviews in Toxicology, vol. 32, no. 3, pp. 211-232, 2002.
[72] R. Blomhoff and H. K. Blomhoff, "Overview of retinoid metabolism and function," Journal ofNeurobiology, vol. 66, no. 7, pp. 606-630, 2006.
[73] G.J. Fisher, S. Datta, Z. Wang et al., "c-Jun-dependent inhibition of cutaneous procollagen transcription following ultraviolet irradiation is reversed by all-trans retinoic acid," Journal of Clinical Investigation, vol. 106, no. 5, pp. 663-670, 2000.
[74] T. T. Schug, D. C. Berry, N. S. Shaw, S. N. Travis, and N. Noy, "Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors," Cell, vol. 129, no. 4, pp. 723-733, 2007.
[75] M. A. Leo and C. S. Lieber, "New pathway for retinol metabolism in liver microsomes," Journal ofBiological Chemistry, vol. 260, no. 9, pp. 5228-5231, 1985.
[76] K. L. Penniston and S. A. Tanumihardjo, "The acute and chronic toxic effects of vitamin A," American Journal of Clinical Nutrition, vol. 83, no. 2, pp. 191-201, 2006.
[77] D. M. Kochar, "Teratogenic activity of retinoic acid," Acta Pathol Microbiol Scand, vol. 70, pp. 398-404, 1967.
[78] R. K. Miller, A. G. Hendricks, J. L. Mills, H. Hummler, and U. W. Weigand, "Periconceptional vitamin A use: how much is teratogenic?" Reproductive Toxicology, vol. 12, pp. 75-88, 1998.
[79] J. A. Olson, "Vitamin A—functions, dietary requirements and safety in humans" in Present Knowledge in Nutrition, E. E. Ziegler and L. J. Filer Jr., Eds., pp. 109-119, International Life Sciences Institute Press, Washington, DC, USA, 7th edition, 2001.
[80] L. H. Allen and M. Haskell, "Estimating the potential for vitamin A toxicity in women and young children," Journal of Nutrition, vol. 132, no. 9, pp. 2907S-2919S, 2002.
[81] D. Coghlan and N. E. Cranswick, "Complementary medicine and vitamin A toxicity in children," Medical Journal of Australia, vol. 175, no. 4, pp. 223-224, 2001.
[82] Institute of Medicine, Food and Nutrition-Based Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Iron, Molybdenum, Nickel, Silicon, Vanadium, and Zinc, National Academy Press, Washington, DC, USA, 2001.
[83] A. Vahlquist, "Vitamin A in human skin: I. Detection and identification of retinoids in normal epidermis," Journal of Investigative Dermatology, vol. 79, no. 2, pp. 89-93, 1982.
[84] A. Vahlquist, J. B. Lee, G. Michaelsson, and O. Rollman, "Vitamin A in human skin: II. Concentrations of carotene, retinol and dehydroretinol in various components of normal skin," Journal ofInvestigative Dermatology, vol. 79, no. 2, pp. 94-97, 1982.
[85] G. Tang, A. R. Webb, R. M. Russell, and M. F. Holick, "Epidermis and serum protect but not retinyl esters from sunlight-induced photodegradation," Photodermatology Photoimmunol-ogy and Photomedicine, vol. 10, no. 1, pp. 1-7, 1994.
[86] M. A. Pathak and T. B. Fitzpatrick, "Preventive treatment of sunburn, dermatoheliosis, and skin cancer with sun-protective agents," in Dermatology in General Medicine, T. Fitzpatrick, A. Z. Eisen, K. Wolff, I. M. Freedberg, K. F. Austen et al., Eds., vol. 1-2, chapter 137, pp. 1689-1716, McGraw-Hill, New York, NY, USA, 4th edition, 1993.
[87] H. F. DeLuca, H. M. Zile, and P. F. Neville, in Lipid Chromatographic Analysis, A. P. Marinette-Guido, Ed., vol. 2, pp. 345-349, Marcel Dekker, New York, NY, USA, 1979.
[88] M. A. Everett, E. Yeargers, R. M. Sayre, and R. L. Olson, "Penetration of epidermis by ultraviolet rays," Photochemistry and Photobiology, vol. 5, no. 7, pp. 533-542, 1966.
[89] J. A. Parrish et al., UV-A. Biological Effects of Ultraviolet Radiation with Emphasis on Human Response to Longwave Ultraviolet, Plenum, New York, NY, USA, 1978.
[90] G. H. Findlay and L. W. van der Merwe, "Epidermal vitamin A and sunburn in man," British Journal ofDermatology, vol. 77, no. 12, pp. 622-626, 1965.
[91] E. Andersson, I. Rosdahl, H. Torma, and A. Vahlquist, "Ultraviolet irradiation depletes cellular retinol and alters the metabolism of retinoic acid in cultured human keratinocytes and melanocytes," Melanoma Research, vol. 9, no. 4, pp. 339-346, 1999.
[92] I. Clark and C. A. Bassett, "The amelioration of hypervitaminosis D in rats with vitamin A," The Journal of Experimental Medicine, vol. 115, pp. 147-156, 1962.
[93] R. Sanchez-Martinez, A. I. Castillo, A. Steinmeyer, and A. Aranda, "The retinoid X receptor ligand restores defective signalling by the vitamin D receptor," EMBO Reports, vol. 7, no. 10, pp. 1030-1034, 2006.
[94] C. Masterjohn, "Vitamin D toxicity redefined: vitamin K and the molecular mechanism," Medical Hypotheses, vol. 68, no. 5, pp. 1026-1034, 2007.
[95] A. F. Morgan, L. Kimmel, and N. C. Hawkins, "A comparison of the hypervitaminosis induced by irradiated ergosterol and ish liver oil concentrates," Te Journal ofBiological Chemistry, vol. 120, no. 1,pp. 85-102, 1937.
[96] A. Aburto, H. M. Edwards Jr., and W. M. Britton, "The influence of vitamin a on the utilization and amelioration of toxicity of cholecalciferol, 25-hydroxycholecalciferol, and 1,25 dihydroxycholecalciferol in young broiler chickens," Poultry Science, vol. 77, no. 4, pp. 585-593, 1998.
[97] A. Aburto and W. M. Britton, "Effects of different levels of vitamins A and E on the utilization of cholecalciferol by broiler chickens," Poultry Science, vol. 77, no. 4, pp. 570-577, 1998.
[98] A. M. Myhre, M. H. Carlsen, S. K. B0hn, H. L. Wold, P. Laake, and R. Blomhoff, "Water-miscible, emulsified, and solid forms of retinol supplements are more toxic than oil-based preparations," American Journal ofClinical Nutrition, vol. 78, no. 6, pp. 1152-1159,2003.
[99] D. Feskanich, V. Singh, W. C. Willett, and G. A. Colditz, "Vitamin A intake and hip fractures among postmenopausal women," Journal ofthe American Medical Association, vol. 287, no. 1, pp. 47-54, 2002.
[100] D. K. Dhanwal, C. Cooper, E. M. Dennison et al., "Geographic variation in osteoporotic hip fracture incidence: the growing importance ofAsian influences in coming decades," Journal of Osteoporosis, vol. 2010, Article ID 757102, 5 pages, 2010.
[101] S. Johansson and H. Melhus, "Vitamin A antagonizes calcium response to vitamin D in man," Journal ofBone and Mineral Research, vol. 16, no. 10, pp. 1899-1905, 2001.
[102] C. M. Rohde and H. F. DeLuca, "All-trans retinoic acid antagonizes the action of calciferol and its active metabolite, 1,25-dihydroxycholecalciferol, in rats," Journal ofNutrition, vol. 135, no. 7, pp. 1647-1652, 2005.
[103] K. Oh, W. C. Willett, K. Wu, C. S. Fuchs, and E. L. Giovannucci, "Calcium and vitamin D intakes in relation to risk ofdistal col-orectal adenoma in women," American Journal of Epidemiology, vol. 165, no. 10, pp. 1178-1186, 2007.
[104] J. S. Adams and B. W. Hollis, "Vitamin D synthesis, metabolism, and clinical measurement," in Diseases ofBone and Mineral Metabolism, F. L. Coe and M. J. Favus, Eds., p. 159, Lippincott Williams & Wilkins, Philadelphia, Pa, USA, 2002.
[105] R. P. Heaney, "Te Vitamin D requirement in health and disease," Journal ofSteroid Biochemistryand Molecular Biology, vol. 97, no. 1-2, pp. 13-19, 2005.
[106] H. M. Page, E. S. Erwin, and G. E. Nelms, "Effect of heat and solar radiation on vitamin A utilization by the bovine animal," American Journal ofPhysiology, vol. 196, pp. 917-918, 1959.
[107] H. K. Biesalski and D. Nohr, "New aspects in vitamin A metabolism: the role of retinyl esters as systemic and local sources for retinol in mucous epithelia," Journal ofNutrition, vol. 134, supplement 12, pp. 3453S-3457S, 2004.
[108] M. Maden and M. Hind, "Retinoic acid in alveolar development, maintenance and regeneration," Philosophical Transactions of the Royal Society, vol. 359, no. 1445, pp. 799-808, 2004.
[109] Y. P. Di, R. Harper, Y. Zhao, N. Pahlavan, W. Finkbeiner, and R. Wu, "Molecular cloning and characterization of spurt, a human novel gene that is retinoic acid-inducible and encodes a secretory protein specific in upper respiratory tracts," Journal ofBiological Chemistry, vol. 278, no. 2, pp. 1165-1173, 2003.
[110] Morbidity and Mortality Weekly Reports, "Prevention and control of influenza," Recommendations of the Advisory Committee on Immunization Practices (ACIP) 53, pp. 1-44, Morbidity and Mortality Weekly Report, 2004.
[111] N. K. Polakos, J. C. Cornejo, D. A. Murray et al., "Kupffer call-dependent hepatitis occurs during influenza infection," The American Journal of Pathology, vol. 168, pp. 1169-1178, 2006.
[112] D. H. Adams and S. G. Hubscher, "Systemic viral infections and collateral damage in the liver," American Journal of Pathology, vol. 168, no. 4, pp. 1057-1059, 2006.
[113] J. R. Whitworth, C. L. Mack, J. A. O'Connor, M. R. Narkewicz, S. Mengshol, and R. J. Sokol, "Acute hepatitis and liver failure associated with influenza a infection in children," Journal of Pediatric Gastroenterology and Nutrition, vol. 43, no. 4, pp. 536-538, 2006.
[114] S. R. Frankel, A. Eardley, G. Lauwers, M. Weiss, and R. P. Warrell, "The "retinoic acid syndrome" in acute promyelocytic leukemia," Annals of Internal Medicine, vol. 117, no. 4, pp. 292-296, 1992.
[115] S. Sacchi, H. M. Kantarjian, E. J. Freireich et al., "Unexpected high incidence of severe toxicities associated with alpha interferon, low-dose cytosine arabinoside and all-trans retinoic acid in patients with chronic myelogenous leukemia," Leukemia and Lymphoma, vol. 35, no. 5-6, pp. 483-489, 1999.
[116] R. Costello, R. Bouabdallah, and J.-A. Gastaut, "Pseudo-"acid retinoic syndrome" mimicked by severe influenza A infection," American Journal of Hematology, vol. 52, p. 120, 1996.
[117] L. E. Davis, M. Kornfeld, R. S. Daniels, and J. J. Skehel, "Experimental influenza causes a non-permissive viral infection of brain, liver and muscle," Journal of NeuroVirology, vol. 6, no. 6, pp. 529-536, 2000.
[118] D. M. Aboulafia, D. Norris, D. Henry et al., "9-cis-retinoic acid capsules in the treatment of AIDS-related Kaposi sarcoma: results of a phase 2 multicenter clinical trial," Archives of Dermatology, vol. 139, no. 2, pp. 178-186, 2003.
[119] C. Sandrock and T. Kelly, "Clinical review: update of avian influenza A infections in humans," Critical Care, vol. 11, no. 2, article 209, 2007.
[120] M. J. Fredette, G. de Serres, and M. Malenfant, "Ophthalmological and biological features ofthe oculorespiratorysyndrome after influenza vaccination," Clinical Infectious Diseases, vol. 37, no. 8, pp. 1136-1138,2003.
[121] F. T. Fraunfelder, F. W. Fraunfelder, and R. Edwards, "Ocular side effects possibly associated with isotretinoin usage," American Journal of Ophthalmology, vol. 132, no. 3, pp. 299-305,2001.
[122] R. D. Semba, "Vitamin A as "anti-infective" therapy, 1920-1940," Journal of Nutrition, vol. 129, no. 4, pp. 783-791, 1999.
[123] S. K. Stansfield, M. Pierre-Louis, G. Lerebours, and A. Augustin, "Vitamin A supplementation and increased prevalence of child-hood diarrhoea and acute respiratory infections," The Lancet, vol. 342, no. 8871, pp. 578-582, 1993.
[124] I. Grotto, M. Mimouni, M. Gdalevich, and D. Mimouni, "Vitamin A supplementation and childhood morbidity from diarrhea and respiratory infections: a meta-analysis," Journal of Pediatrics, vol. 142, no. 3, pp. 297-304, 2003.
[125] C. B. Stephensen, L. M. Franchi, H. Hernandez, M. Campos, R. H. Gilman, and J. O. Alvarez, "Adverse effects of high-dose vitamin A supplements in children hospitalized with pneumonia," Pediatrics, vol. 101, no. 5, p. E3, 1998.
[126] D. Cui, Z. Moldoveanu, and C. B. Stephensen, "High-level dietary vitamin A enhances T-helper type 2 cytokine production and secretory immunoglobulin A response to influenza A virus infection in BALB/c mice," Journal of Nutrition, vol. 130, no. 5, pp. 1132-1139,2000.
[127] H. Chen, Q. Zhuo, W. Yuan, J. Wang, and T. Wu, "Vitamin A for preventing acute lower respiratory tract infections in children up to seven years of age," Cochrane Database of Systematic Reviews, no. 1, article CD006090, 2008.
[128] J. Ni, J. Wei, and T. Wu, "Vitamin A for non-measles pneumonia in children," Cochrane Database of Systematic Reviews, no. 3, p. CD003700, 2005.
[129] P. Ghazal, J. LeBlanc, and A. Angulo, "Vitamin A regulation of viral growth," Reviews in Medical Virology, vol. 7, pp. 21-34,1997.
[130] D. Wolff, C. Sinzger, P. Drescher, G. Jahn, and B. Plachter, "Reduced levels of IE2 gene expression and shutdown of early and late viral genes during latent infection ofthe glioblastoma cell line U138-MG with selectable recombinants of human cytomegalovirus," Virology, vol. 204, no. 1, pp. 101-113, 1994.
[131] P. Ghazal, C. DeMattei, E. Giulietti, S. A. Kliewer, K. Umesono, and R. M. Evans, "Retinoic acid receptors initiate induction of the cytomegalovirus enhancer in embryonal cells," Proceedings of the National Academy of Sciences of the United States of America, vol. 89, no. 16, pp. 7630-7634, 1992.
[132] A. Angulo, R. A. R. Chandraratna, J. F. LeBlanc, and P. Ghazal, "Ligand induction of retinoic acid receptors alters an acute infection by murine cytomegalovirus," Journal ofVirology, vol. 72, no. 6, pp. 4589-4600, 1998.
[133] J. A. Turpin, M. Vargo, and M. S. Meltzer, "Enhanced HIV-1 replication in retinoid-treated monocytes: retinoid effects mediated through mechanisms related to cell differentiation and to a direct transcriptional action on viral gene expression," Journal of Immunology, vol. 148, no. 8, pp. 2539-2546, 1992.
[134] M. O. Lee, P. D. Hobbs, X. K. Zhang, M. I. Dawson, and M. Pfahl, "A synthetic retinoid antagonist inhibits the human immunodeficiency virus type 1 promotor," Proceedings ofthe National Academy of Sciences of the United States of America, vol. 91, pp. 5632-5636, 1994.
[135] R. J. Jones, S. Dickerson, P. M. Bhendaet al., "Epstein-Barr virus lytic infection induces retinoic acid-responsive genes through of a retinol-metabolizing enzyme, DHRS9," Journal of Biological Chemistry, vol. 282, no. 11, pp. 8317-8324, 2007.
[136] T. Ichinohe, A. Iwasaki, and H. Hasegawa, "Innate sensors of influenza virus: clues to developing better intranasal vaccines" Expert Review of Vaccines, vol. 7, no. 9, pp. 1435-1445, 2008.
[137] A. Iwasaki and R. Medzhitov, "Toll-like receptor control of the adaptive immune responses," Nature Immunology, vol. 5, no. 10, pp. 987-995, 2004.
[138] J. M. Lund, L. Alexopoulou, A. Sato et al., "Recognition of single-stranded RNA viruses by Toll-like receptor 7," Proceedings ofthe National Academy ofSciences ofthe United States of America, vol. 101, no. 15, pp. 5598-5603, 2004.
[139] B. Pulendran, "Tolls and beyond—many roads to vaccine immunity," New England Journal ofMedicine, vol. 356, no. 17, pp. 1776-1778,2007.
[140] J. Siren, T. Imaizumi, D. Sarkar et al., "Retinoic acid inducible gene-I and mda-5 are involved in influenza A virus-induced expression of antiviral cytokines," Microbes and Infection, vol. 8, no. 8, pp. 2013-2020, 2006.
[141] H. Kato, S. Sato, M. Yoneyama et al., "Cell type-specific involvement of RIG-I in antiviral response" Immunity, vol. 23, no. 1,pp. 19-28,2005.
[142] S. S. Diebold, T. Kaisho, H. Hemmi, S. Akira, and C. R. E. Sousa, "Innate antiviral responses by means of TLR7-mediated recognition ofsingle-stranded RNA," Science, vol. 303, no. 5663, pp. 1529-1531,2004.
[143] T. Hennet, H. J. Ziltener, K. Frei, and E. Peterhans, "A kinetic study ofimmune mediators in the lungs of mice infected with influenza A virus," Journal of Immunology, vol. 149, no. 3, pp. 932-939, 1992.
[144] J. Pirhonen, T. Sareneva, M. Kurimoto, I. Julkunen, and S. Matikainen, "Virus infection activates IL-1/5 and IL-18 production in human macrophages by a caspase-1-depend-ent pathway," Journal of Immunology, vol. 162, no. 12, pp. 7322-7329, 1999.
[145] N. Schmitz, M. Kurrer, M. F. Bachmann, and M. Kopf, "Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection" Journal of Virology, vol. 79, no. 10, pp. 6441-6448, 2005.
[146] J. D. Fontenot, M. A. Gavin, and A. Y. Rudensky, "Foxp3 programs the development and function of CD4+CD25+ regulatory T cells" Nature Immunology, vol. 4, no. 4, pp. 330-336, 2003.
[147] J. Dai, B. Liu, and Z. Li, "Regulatory T cells and Toll-like receptors: what is the missing link?" International Immuno-pharmacology, vol. 9, no. 5, pp. 528-533, 2009.
[148] C. M. Sun, J. A. Hall, R. B. Blank et al., "Small intestine lamina propria dendritic cells promote de novo generation ofFoxp3 T reg cells via retinoic acid" Journal of Experimental Medicine, vol. 204, no. 8, pp. 1775-1785, 2007.
[149] J. L. Coombes, K. R. R. Siddiqui, C. V. Arancibia-Carcamo et al., "A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-/5 and retinoic acid-dependent mechanism," Journal ofExperimental Medicine, vol. 204, no. 8, pp. 1757-1764, 2007.
[150] S. Uematsu, K. Fujimoto, M. H. Jang et al., "Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5," Nature Immunology, vol. 9, no. 7, pp. 769-776, 2008.
[151] C. H. Kim, "Regulation of FoxP3+ regulatory T cells and Th17 cells by retinoids" Clinical and Developmental Immunology, vol. 2008, Article ID 416910, 12 pages, 2008.
[152] D. Mucida, Y. Park, G. Kim et al., "Reciprocal TT17 and regulatory T cell differentiation mediated by retinoic acid" Science, vol. 317, no. 5835, pp. 256-260, 2007.
[153] M. Iwata, A. Hirakiyama, Y. Eshima, H. Kagechika, C. Kato, and S. Y. Song, "Retinoic acid imprints gut-homing specificity on T cells," Immunity, vol. 21, no. 4, pp. 527-538, 2004.
[154] J. F. Bermejo-Martin, I. Martin-Loeches, J. Rello et al., "Host adaptive immunity deficiency in severe pandemic influenza," Critical Care, vol. 14, no. 5, R167, 2010.
[155] J. Louten, K. Boniface, and R. de Waal Malefyt, "Development and function of TH17 cells in health and disease," Journal of Allergy and Clinical Immunology, vol. 123, no. 5, pp. 1004-1011, 2009.
[156] G. Krstic, "Th17 mediators and vitamin D status" Critical Care, vol. 14, no. 2, article 410, 2010.
[157] J. P. Wang, G. N. Bowers, C. Padden et al., "Toll-like receptor-mediated activation of neutrophils by influenza virus" Blood, vol. 112, pp. 2028-2034, 2008.
[158] M. D. de Jong, C. P. Simmons, T. T. Thanh et al., "Fatal outcome of human influenza A (H5N1) is associated wth high viral-load and hypercytokinemia" Nature Medicine, vol. 12, no. 10, pp. 1203-1207, 2006.
[159] N. D. Lawson and N. Berliner, "Neutrophil maturation and the role of retinoic acid," Experimental Hematology, vol. 27, pp. 1355-1367, 1999.
[160] K. Mehta, "Retinoic acid—a player that rules the game of life and death in neutrophils," Indian Journal ofExperimental Biology, vol. 40, no. 8, pp. 874-881, 2002.
[161] K. A. Daher, M. E. Selsted, and R. I. Lehrer, "Directinactivation of viruses by human granulocyte defensins" Journal of Virology, vol. 60, no. 3, pp. 1068-1074, 1986.
[162] R. I. Lehrer, "Multispecific myeloid defensins" Current Opinion in Hematology, vol. 14, no. 1, pp. 16-21, 2007.
[163] H. Koga, I. Fujita, and S. Miyazaki, "Effects of all-trans-retinoic acid on superoxide generation in intact neutrophils and a cell-free system," British Journal of Haematology, vol. 97, no. 2, pp. 300-305, 1997.
[164] S. S. Twining, D. P. Schulte, P. M. Wilson, B. L. Fish, and J. E. Moulder, "Vitamin A deficiency alters rat neutrophil function" Journal of Nutrition, vol. 127, no. 4, pp. 558-565, 1997.
[165] J. Jagdeo, R. Campbell, T. Long, J. Muglia, G. Telang, and L. Robinson-Bostom, "Sweet's syndrome-like neutrophilic lobular panniculitis associated with all-trans-retinoic acid chemotherapy in a patient with acute promyelocytic leukemia," Journal of the American Academy of Dermatology, vol. 56, no. 4, pp. 690-693, 2007.
[166] Y. Zhao, M. Lu, L. T. Lau et al., "Neutrophils may be a vehicle for viral replication and dissemination in human H5N1 avian influenza," Clinical Infectious Diseases, vol. 47, no. 12, pp. 1575-1578,2008.
[167] L. Kaiser, R. S. Fritz, S. E. Straus, L. Gubareva, and F. G. Hayden, "Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses," Journal of Medical Virology, vol. 64, no. 3, pp. 262-268, 2001.
[168] M. L. Heltzer, S. E. Coffin, K. Maurer et al., "Immune dysregulation in severe influenza," Journal of Leukocyte Biology, vol. 85, no. 6, pp. 1036-1043, 2009.
[169] N. Krishnamoorthy, T. B. Oriss, M. Paglia et al., "Activation of c-Kit in dendritic cells regulates T helper cell differentiation and allergic asthma," Nature Medicine, vol. 14, no. 5, pp. 565-573, 2008.
[170] M. Iwata, Y. Eshima, and H. Kagechika, "Retinoic acids exert direct effects on T cells to suppress Th1 development and enhance T1 development via retinoic acid receptors," International Immunology, vol. 15, no. 8, pp. 1017-1025, 2003.
[171] J. R. Mora, M. Iwata, B. Eksteen et al., "Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells," Science, vol. 314, no. 5802, pp. 1157-1160, 2006.
[172] H. Asano, M. Aonuma, T. Sanosaka, J. Kohyama, M. Nami-hira, and K. Nakashima, "Astrocyte differentiation of neural precursor cells is enhanced by retinoic acid through a change in epigenetic modification," Stem Cells, vol. 27, no. 11, pp. 2744-2752, 2009.
[173] F. Herrera, Q. Chen, and D. Schubert, "Synergistic effect of retinoic acid and cytokines on the regulation of glial ibrillary acidic protein expression," Journal of Biological Chemistry, vol. 285, no. 50, pp. 38915-38922, 2010.
[174] S. Oh, J. M. Mccaffery, and M. C. Eichelberger, "Dose-dependent changes in influenza virus-infected dendritic cells result in increased allogeneic T-cell proliferation at low, but not high, doses of virus," Journal of Virology, vol. 74, no. 12, pp. 5460-5469, 2000.
[175] T. P. Welliver, J. L. Reed, and R. C. Welliver Sr., "Respiratory syncytial virus and influenza virus infections: observations from tissues of fatal infant cases," The Pediatric Infectious Disease Journal, vol. 27, no. 10, pp. S92-S96, 2008.
[176] D. Proud, "Nitric oxide and the common cold," Current Opinion in Allergy and Clinical Immunology, vol. 5, no. 1, pp. 37-42, 2005.
[177] C. Watanabe, H. Kawashima, K. Takekuma, A. Hoshika, and Y. Watanabe, "Increased nitric oxide production and GFAP expression in the brains of influenza A/NWS virus infected mice," Neurochemical Research, vol. 33, no. 6, pp. 1017-1023, 2008.
[178] D. Personett, U. Pass, K. Panickar, and M. McKinney, "Retinoic acid-mediated enhancement of the cholinergic/neuronal nitric oxide synthase phenotype of the medial septal SN56 clone: establishment of a nitric oxide-sensitive proapoptotic state," Journal of Neurochemistry, vol. 74, no. 6, pp. 2412-2424, 2000.
[179] S. Moncada, R. M. J. Palmer, and E. A. Higgs, "Nitric oxide: physiology, pathophysiology, and pharmacology," Pharmacological Reviews, vol. 43, no. 2, pp. 109-142, 1991.
[180] K. Mehta, T. McQueen, S. Tucker, R. Pandita, and B. B. Aggar-wal, "Inhibition of all-trans-retinoic acid of tumor necrosis factor and nitric oxide production by peritoneal macrophages" Journal of Leukocyte Biology, vol. 55, no. 3, pp. 336-342, 1994.
[181] K. Hirokawa, K. M. O'Shaughnessy, P. Ramrakha, and M. R. Wilkins, "Inhibition of nitric oxide synthesis in vascular smooth muscle by retinoids," British Journal of Pharmacology, vol. 113, no. 4, pp. 1448-1454, 1994.
[182] K. Motomura, H. Sakai, H. Isobe, and H. Nawata, "Effects of retinoids on the production of tumour necrosis factor-alpha and nitric oxide by lipopolysaccharide stimulated rat Kupffer cells in vitro: evidence for participation of retinoid X receptor signalling pathway" Cell Biochemistry and Function, vol. 15, no. 2, pp. 95-101, 1997.
[183] A. Sirsjo, A. C. Gidlof, A. Olsson et al., "Retinoic acid inhibits nitric oxide synthase-2 expression through the retinoic acid receptor-oc," Biochemical and Biophysical Research Communications, vol. 270, no. 3, pp. 846-851, 2000.
[184] A. Duchini, M. E. Viernes, L. M. Nyberg, R. M. Hendry, and P. J. Pockros, "Hepatic decompensation in patients with cirrhosis during infection with influenza A," Archives of Internal Medicine, vol. 160, no. 1, pp. 113-115, 2000.
[185] A. B. Barua, P. K. Duitsman, D. Kostic, M. Barua, and J. A. Olson, "Reduction of serum retinol levels following a single oral dose of all-trans retinoic acid in humans" International Journal for Vitamin and Nutrition Research, vol. 67, no. 6, pp. 423-426, 1997.
[186] C. S. Lieber and M. A. Leo, "Interaction of alcohol and nutritional factors with hepatic fibrosis," Progress in Liver Diseases, vol. 8, pp. 253-272, 1986.
[187] Z. Dan, Y. Popov, E. Patsenker et al., "Hepatotoxicity of alcohol-induced polar retinol metabolites involves apoptosis via loss of mitochondrial membrane potential," The FASEB Journal, vol. 19, no. 7, pp. 845-847, 2005.
[188] W. Sim, E. Abril, and D. Earnest, "Mechanisms of Kupffer cell activation in hypervitaminosis A," in Cells of the Hepatic Sinusoid, E. Wisse, D. Knook, and K. Decker, Eds., pp. 91-93, The Kupffer Cell Foundation, Rijswijk, The Netherlands, 1989.
[189] S. A. Mobley, W. Sim, and I. G. Sipes, "Repeated exposure to subclinical doses of hepatotoxicant enhances liver injury with hypervitaminosis A: a possible cause for chronic liver disease," Gastroenterology, vol. 96, p. A689, 1989.
[190] R. M. Russell, J. L. Boyer, S. A. Bagheri, and Z. Hruban, "Hepatic injury from chronic hypervitaminosis A resulting in portal hypertension and ascites," New England Journal of Medicine, vol. 291, no. 9, pp. 435-440, 1974.
[191] R. Cheruvattath, M. Orrego, M. Gautam et al., "Vitamin A toxicity: when one a day doesn't keep the doctor away," Liver Transplantation, vol. 12, no. 12, pp. 1888-1891, 2006.
[192] V. S. Ramanathan, G. Hensley, S. French et al., "Hypervitaminosis A inducing intra-hepatic cholestasis—a rare case report," Experimental and Molecular Pathology, vol. 88, no. 2,pp. 324-325, 2010.
[193] D. Li and S. L. Friedman, "Liver ibrogenesis and the role of hepatic stellate cells: new insights and prospects for therapy," Journal of Gastroenterology and Hepatology, vol. 14, no. 7, pp. 618-633, 1999.
[194] M. Okuno, S. Kojima, K. Akita et al., "Retinoids in liver fibrosis and cancer," Front Biosci, vol. 7, pp. d204-d218, 2002. *[195] J. Mann and D. A. Mann, "Transcriptional regulation of hepatic stellate cells," Advanced Drug Delivery Reviews, vol. 61, no. 7-8, pp. 497-512,2009. *[196] H. Popper and F. Schaffner, "Cholestasis," in Gastroenterology, J. E. Berk, Ed., vol. 5, chapter 148, pp. 2697-2731, WB Saunders, Philadelphia, Pa, USA, 4th edition, 1985.
[197] C. A. Frolik, "Metabolism of retinoids," in The Retinoids, M. B. Sporn, A. B. Roberts, and D. S. Goodman, Eds., pp. 177-208, Academic Press, New York, NY, USA, 1984.
[198] V. A. Hicks, D. B. Gunning, and J. A. Olson, "Metabolism, plasma transport and biliary excretion of radioactive vitamin A and its metabolites as a function of liver reserves of vitamin Aintherat," Journal ofNutrition, vol. 114, no. 7,pp. 1327-1333, 1984.
[199] A. K. Mitra, J. O. Alvarez, M. A. Wahed, G. J. Fuchs, and C. B. Stephensen, "Predictors of serum retinol in children with shigellosis," American Journal ofClinical Nutrition, vol. 68, no. 5, pp. 1088-1094, 1998.
[200] F. J. Schweigert, "inflammation-induced changes in the nutritional biomarkers serum retinol and carotenoids," Current Opinion in Clinical Nutrition and Metabolic Care, vol. 4, no. 6, pp. 477-481,2001.
[201] S. H. Gieng, M. H. Green, J. B. Green, and F. J. Rosales, "Model-based compartmental analysis indicates a reduced mobilization of hepatic vitamin A during inflammation in rats," Journal of
Lipid Research, vol. 48, no. 4, pp. 904-913, 2007.
[202] E. Mezey, "Liver disease and protein needs," Annual Review of Nutrition, vol. 2, pp. 21-50, 1982.
[203] T. R. Martin and C. W. Frevert, "Innate immunity in the lungs," Proceedings ofthe American Toracic Society, vol. 2, no. 5, pp. 403-411,2005.
[204] T. M. Tumpey, X. Lu, T. Morken, S. R. Zaki, and J. M. Katz, "Depletion of lymphocytes and diminished cytokine production in mice infected with a highly virulent influenza A (H5N1) virus isolated from humans," Journal of Virology, vol. 74, no. 13, pp. 6105-6116, 2000.
[205] I. A. Clark, L. M. Alleva, and B. Vissel, "The roles of TNF in brain dysfunction and disease," Pharmacology & Therapeutics, vol. 128, no. 3, pp. 519-548, 2010.
[206] T. Gustot, F. Durand, D. Lebrec, J. L. Vincent, and R. Moreau, "Severe sepsis in cirrhosis," Hepatology, vol. 50, no. 6, pp. 2022-2033, 2009.
[207] A. Ukleja, J. S. Scolapio, J. P. McConnell et al., "Nutritional assessment of serum and hepatic vitamin A levels in patients with cirrhosis," Journal ofParenteral and Enteral Nutrition, vol. 26, no. 3, pp. 184-188,2002.
[208] H. Maeda and T. Akaike, "Nitric oxide and oxygen radicals in infection, inflammation and cancer," Biochemistry, vol. 63, no. 7, pp. 854-865, 1998.
[209] D. Ungheri, C. Pinasi, G. Sanson et al., "Protective effect of N-acetylcysteine in a model of influenza infection in mice" International Journal of Immunopathology and Pharmacology, vol. 13, no. 3, pp. 123-128, 2000.
[210] R. Harrison, "Structure and function of xanthine oxidoreduc-tase: where are we now?" Free Radical Biology and Medicine, vol. 33, no. 6, pp. 774-797, 2002.
[211] G. Taibi, A. Paganini, M. C. Gueli, F. Ampola, and C. M. A. Nicotra, "Xanthine oxidase catalyzes the synthesis of retinoic acid" Journal of Enzyme Inhibition, vol. 16, no. 3, pp. 275-285, 2001.
[212] A. Kumar, R. Zarychanski, R. Pinto et al., "Hospitalized patients with 2009 H1N1 influenza in the United States, April-June 2009," The New England Journal of Medicine, vol. 361, pp. 1935-1944, 2009.
[213] J. F. Bermejo-Martin, R. Ortiz de Lejarazu, T. Pumarola et al., "Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza" Critical Care, vol. 13, no. 6, p. R201,2009.
[214] Centers for Disease Control and Prevention (CDC), "Intensive-care patients with severe novel influenza A (H1N1) virus infection—Michigan, June 2009," Morbidity and Mortality Weekly Report, vol. 58, no. 27, pp. 749-752, 2009.
[215] C. Venkata, P. Sampathkumar, and B. Afessa, "Hospitalized patients with 2009 H1N1 influenza infection: the mayo clinic experience," Te Mayo Clinic Proceedings, vol. 85, no. 9, pp. 798-805, 2010.
[216] Q. Yang, T. E. Graham, N. Mody et al., "Serum retinol binding protein 4 contributes to insulin resistance in obesityand type 2 diabetes," Nature, vol. 436, no. 7049, pp. 356-362, 2005.
[217] T. E. Graham, Q. Yang, M. Bluher et al., "Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects," New England Journal ofMedicine, vol. 354, no. 24, pp. 2552-2563, 2006.
[218] C. C. Chen, J. Y. Wu, C. T. Chang et al., "Levels of retinol-binding protein 4 and uric acid in patients with type 2 diabetes mellitus," Metabolism, vol. 58, no. 12, pp. 1812-1816, 2009.
[219] C. Erikstrup, O. H. Mortensen, A. R. Nielsen et al., "RBP-to-retinol ratio, but not total RBP, is elevated in patients with type 2 diabetes" Diabetes, Obesity and Metabolism, vol. 11, no. 3, pp. 204-212,2009.
[220] A. R. Mawson, W. Bennett, S. Tanumihardjo et al., "Altered retinoid profiles in an animal model of preeclampsia, gestational diabetes and fetal growth restriction" Reproductive Sciences, vol. 17, supplement 3, abstract 592, p. 235A, 2010.
[221] C. F. Basler, "influenza viruses: basic biologyand potential drug targets," Infectious Disorders, vol. 7, no. 4, pp. 282-293, 2007.
[222] D. S. Fedson, "Confronting an influenza pandemic with inexpensive generic agents: can it be done?" The Lancet Infectious Diseases, vol. 8, no. 9, pp. 571-576, 2008.
[223] J. Bassaganya-Riera, R. Song, P. C. Roberts, and R. Hontecillas, "PPAR-y activation as an anti-inflammatory therapy for respiratory virus infections" Viral Immunology, vol. 23, no. 4, pp. 343-352, 2010.
[224] J. M. Katz, J. Plowden, M. Renshaw-Hoelscheret al., "Immunity to influenza: the challenges of protecting an aging population" Immunology Research, vol. 29, no. 1-3, pp. 113-124, 2004.
[225] P. J. Garry, W. C. Hunt, J. L. Bandrofchak, D. VanderJagt, and J. S. Goodwin, "Vitamin A intake and plasma retinol levels in healthy elderly men and women" American Journal of Clinical Nutrition, vol. 46, no. 6, pp. 989-994, 1987.
[226] G. W. Comstock, M. S. Menkes, S. E. Schober, J. P. Vuilleumier, and K. J. Helsing, "Serum levels of retinol, beta-carotene, and alpha-tocopherol in older adults," American Journal of Epidemiology, vol. 127, no. 1,pp. 114-123, 1988.
[227] D. Sklan, S. Trifon, O. Kedar, N. Vaisman, and Y. Berner, "Retinoid metabolism in human leucocytes," British Journal of Nutrition, vol. 73, no. 6, pp. 889-895, 1995.
[228] D. Hollander and V. Dadufalza, "influence of aging on vitamin A transport into the lymphatic circulation" Experimental Gerontology, vol. 25, no. 1, pp. 61-65, 1990.
[229] S. D. Krasinski, J. S. Kohn, E. J. Schaefer, and R. M. Russell, "Post prandial plasma retinyl ester response is greafter in older subjects compared with younger subjects," Journal of Clinical Investigation, vol. 85, pp. 883-892, 1990.
[230] J. B. Johnson, D. R. Laub, and S. John, "The effect on health of alternate day calorie restriction: eating less and more than needed on alternate days prolongs life" Medical Hypotheses, vol. 67, no. 2, pp. 209-211,2006.
[231] T. Akaike, M. Ando, T. Oda et al., "Dependence on O2-generation by xanthine oxidase of pathogenesis of influenza virus infection in mice," Journal ofClinical Investigation, vol. 85, no. 3, pp. 739-745, 1990.
[232] C. Apfel, F. Bauer, M. Crettaz et al., "A retinoic acid receptor a antagonist selectively counteracts retinoic acid effects," Proceedings of the National Academy of Sciences of the United States of America, vol. 89, no. 15, pp. 7129-7133, 1992.