Interaction of drugs with Vitamin D, Magnesium, Vitamin B12, Selenium, etc
Important drug-micronutrient interactions: A selection for clinical practice.
Crit Rev Food Sci Nutr. 2018 Dec 23:1-19. doi: 10.1080/10408398.2018.1522613.
Gröber U1, Schmidt J1, Kisters K1,2.
1 Academy of Micronutrient Medicine , Essen , Germany.
2 Medizinische Klinik I , St. Anna Hospital , Herne , Germany.

📄 Download the PDF from Sci-Hub via VitaminDWiki
Interactions between drugs and micron3trients have received only little or no attention in the medical and pharmaceutical world in the past. Since more and more pharmaceutics are used for the treatment of patients, this topic is increasingly relevant. As such interactions - depending on the duration of treatment and the status of micronutrients - impact the health of the patient and the action of the drugs, physicians and pharmacists should pay more attention to such interactions in the future. This review aims to sensitize physicians and pharmacists on drug micronutrient interactions with selected examples of widely pescribed drugs that can precipitate micronutrient deficiencies. In this context, the pharmacist, as a drug expert, assumes a particular role. Like no other professional in the health care sector, he is particularly predestined and called up to respond to this task. The following article intends to point out the relevance of mutual interactions between micronutrients and various examples of widely used drugs, without claiming to be exhaustive.
KEYWORDS: Metformin; Micronutrient; coenzyme Q10; drug; interactions; magnesium; proton pump inhibitors; selenium; statins; thiazide diuretics; vitamin B12; vitamin D
Introduction
Thanks to modern health care and the improvement of life quality, the average life expectancy of Europeans has almost doubled over the past 100 years. Consequently in the European Union the demographic old-age dependency ratio continues to rise significantly over the coming decades due to a large increase in the population above 65 years old. Being about 25% in 2010, it has risen to 29.6% in 2016 and is projected to rise further, in particular up to 2050, and eventually reach 51.2% in 2070 (Economic and Financial Affairs 2017). The increase in the mean age is associated with an increasing number of multimorbid patients, who suffer from nutrition-associated diseases and usually depend on complex pharmacotherapy (Hall et al. 2018; Hofer- Diickelmann 2012). For example a population study in Scotland found an overall prevalence of multimorbidity of 23.2% (Barnett et al. 2012). A recent cross-sectional analysis in Germany with more than 10,000 participants aged 50 years and older revealed that even more than 95% of the patients with osteoporosis had at least one coexisting disease (Puth et al. 2018). The prevalence of multimorbidity in a population increases with age and leads inevitably to polypharmacotherapy. Polypharmacotherapy is a major concern in the elderly (Hofer-Duckelmann 2012; Harugeri et al. 2010). 40% of institutionalized patients take more than nine drugs on a daily basis (Dwyer et al. 2010). Each additional medication, however, increases the risk of adverse drug reactions (Smithburger et al. 2015; Grober 2006).
It is suggested that more than 5% of all hospital admissions are the result of adverse drug reactions, and that about 20% of inpatients have at least one adverse event during their hospital stay. The actual incidence of adverse drug reactions may be even greater because some of them present often as symptoms of a disease and may therefore be undetected. As doctors and researchers with clinical practice for more than 25 years involved in the subject of adverse drug reactions, the presented data and studies in pubmed about drug-micronutrient interactions are very scarce. Furthermore many studies explored these interactions using a drug as monotherapy. Moreover, little consideration is often given to individual patients’ experiences and reporting of specific drug-micronutrient interaction. But this does not correspond to usual clinical practice where drugs are frequently combined, exposing patients to potential synergic effects on micronutrient metabolism.
In clinical practice the decision of the physician and/or pharmacist to screen for drug induced micronutrient deficiencies should therefore rely on an individualized approach taking into account the patient’s medication, his comorbidities, his diet, and lifestyle factors (Fig. 1) (Schatz and Weber 2015; Budnitz et al. 2011).
In the year 1998 a meta-analysis of 39 prospective studies from US hospitals has shown that the overall incidence of serious adverse drug reactions was 6.7% (95% CI: 5.2-8.2%) and of fatal adverse drug reactions was 0.32% (95% CI: 0.23-0.41%) of hospitalized patients. In this analysis adverse drug reactions were the fourth and sixth leading cause of death. Although the results of this analysis should not be overrated given the heterogeneity of the conducted studies, it becomes clear that adverse drug reactions are of high clinical relevance and can take on critical dimensions (Lazarou, Pomeranz, and Corey 1998; Kvasz et al. 2000; Miguel et al. 2012).
Micronutrients have major impact on health
Vitamins and other micronutrients have considerable potential in the prevention and treatment of diet-related diseases. In general, micronutrient is the umbrella term used to mark essential vitamins, minerals and trace elements required from the diet to sustain virtually all normal cellular and molecular functions. It is widely recognized that micronutrient deficiencies are a significant public health problem. Chronic micronutrient deficiencies can lead to complex metabolic disorders which, over the years, lay the foundation for serious lifestyle-related illness. The immune system is weakened, leading to an increased prevalence of infectious diseases, and susceptibility to chronic degenerative diseases increases, as physical and mental development and functional capacity in general are clearly reduced (Grober 2009). An estimated 2 billion people worldwide suffer from micronutrient deficiencies. Those commonly affected include schoolchildren, working men and women, pregnant women, sportsmen and sportswomen, the elderly, and people with impaired absorption or who are on regular medication. Chronic illness and multimorbidity, particularly in the elderly, necessitate greater consumption of drugs, which can considerably impair the absorption and utilization of micronutrients. For example, globally one in two preschool children suffers from anemia, many of them because of Iron deficiency. Furthermore, about 190 million preschool children suffer from vitamin A deficiency, but deficiencies in iodine, vitamin D , folate, and zinc are also highly prevalent. Inadequate micronutrient intake can have serious health consequences for individuals, but also has a wider impact on economies, healthcare systems, societies, and welfare systems. A recent publication describes that the use of dietary supplements among those who are at a high risk of experiencing a costly disease-related event can lead to healthcare cost savings. For example, the full utilization of folic acid, B6 , and B12 among the target population at preventive intake level’s effect on potential avoided coronary heart disease (CHD)-related hospital utilization costs would be an average savings of $1.52 billion per year—a cumulative cost avoidance to health care payers of $12.12 billion from 2013 to 2020. The potential net savings in avoided CHD-related health care costs after accounting for the cost of folic acid, B6 , and B12 utilization at preventive daily intake levels would be an average of $654.0 million per year and more than $5.23 billion in cumulative health care cost net savings from 2013 to 2020 (Peter et al. 2014; Shanahan and de Lorimier 2013).
Drugs and micronutrients
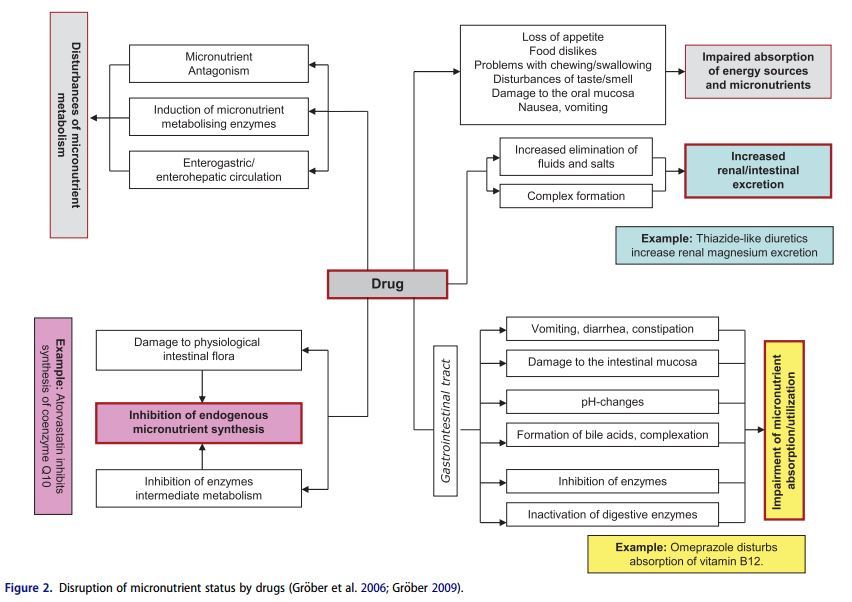
Drugs and micronutrients use the same transport and metabolism pathways in the body for their intestinal absorption, metabolism, and elimination. This means that when one or more drugs are taken, there is always a potential risk of interactions with the nutrient status. Consequently the action of a drug may be adversely affected by a micronutrient (e.g. calcium, Magnesium and zinc can interfere with the gastrointestinal absorption of tetracycline antibiotics) and simultaneously the physiological function of a vitamin or mineral may be impaired by a drug (e.g. methotrexate is a folic acid antagonist that can cause mucosal, gastrointestinal, hepatic or hematologic side effects) (Brion, Lambs, and Berthon 1985; Shea et al. 2014). Disruption of micronutrient status can result in serious metabolic dysfunctions, as there is hardly a single physiological process in the body that is not mediated by one or other of these biocatalysts. Given the ever-increasing number of drugs on the market and the frequency with which they are used, greater attention must be paid in daily medical and pharmaceutical practice focused in particular on the adverse effects of drug therapy on the micronutrient status (Fig. 2) in order to minimize the potential risk to the health of patients. This review aims to sensitize physicians and pharmacists on important drug micronutrient interactions with selected examples of widely pescribed drugs that can precipitate micronutrient deficiencies.
Proton pump inhibitors (PPIs)
First introduced in 1989, proton pump inhibitors (PPIs) are among the most widely utilized medications worldwide, both in the ambulatory and inpatient clinical settings, with a high inappropriate prescription rates, exceeding partially 50% in the elderly (Parsons et al. 2012; Zhang et al. 2017; Boucherie et al. 2018). PPIs block the gastric H+/K+- ATPase, inhibiting gastric acid secretion. This effect enables healing of peptic ulcers, gastroesophageal reflux disease (GERD), Barrett’s esophagus, and Zollinger-Ellison syndrome, as well as the eradication of Helicobacter pylori as part of combination regimens. But by increasing the intra- gastric pH, PPIs can impair the absorption and utilization of micronutrients such as Iron , calcium, Magnesium , vitamin C and vitamin B12 . This can induce clinical problems related to the deficiencies of these micronutrients (e.g. anemia, fractures, hypomagnesemia, vitamin B12 deficiency) (Ito and Jensen 2010).
PPIs and vitamin B12
Vitamin B12 (Cobalamins) is an essential water-soluble vitamin that is vitally important in hematopoiesis, nervous system functions, maintenance of intact gastrointestinal mucosa and regulation of numerous other B12 -dependent metabolic processes. Cobalamins contributes significantly to hematopoiesis, myelin synthesis and synthesis of epithelial tissue. As a coenzyme, it is also a principal component of fatty acid, carbohydrate and nucleic acid metabolism.
The metabolic pathway of the active absorption of vitamin B12 starts when dietary cobalamin (Cbl), obtained through animal foods, enters the stomach bound to animal proteins. Hydrochloric acid (HCl) and Pepsin in the stomach serve the animal protein, releasing free cobalamin. Most of the free cobalamin is then bound to R-protein (R), which is released from the parietal and salivary cells. Intrinsic factor (IF) is also secreted in the stomach, but its binding to cobalamin is weak in the presence of gastric and salivary R- protein. In the duodenum, dietary cobalamin bound to R- protein is joined by cobalamin-R-protein complexes that have been secreted in the bile. Pancreatic enzymes degrade both biliary and dietary cobalamin-R-protein complexes, releasing free cobalamin. The cobalamin then binds with intrinsic factor. The cobalamin-intrinsic factor complex remains undisturbed until the distal 80 cm of the ileum, where it attaches to mucosal cell receptors (cubilin) and the cobalamin is bound to transport proteins known as transco- balamin I, II and III (TCI, TCII and TCIII). Transcobalamin II, although it represents only a small fraction (about 10%) of the transcobalamins, is the most important because it is able to deliver cobalamin to all cells in the body.
The cobalamin is subsequently transported systemically via the portal system. Within each cell, the transcobalamin Il-cobalamin complex is taken up by means of endocytosis and the cobalamin is liberated and then converted enzymatically into its 2 coenzyme forms, methylcobalamin and adenosylcobalamin. Vitamin B12 absorption can also take place independently of the intrinsic factor by means of passive diffusion in the small intestine. This mechanism, however, is not very efficient, since only about 1% of the ingested Vitamin B12 dose is absorbed (Festen 1991; Andres et al. 2004; Grober, Kisters, and Schmidt 2013).
The increase of intragastric pH by PPIs disturbs the active absorption of vitamin B12 by limiting the vitamins cleavage from dietary proteins. Reduced gastric acid production in the stomachs of older persons leads to alkalizing of the small intestine milieu, which eliminates the physiological barrier to microorganisms. Bacteria from lower intestinal segments can then increasingly enter the jejunum and ileum. Bacterial overgrowth with Clostridia and Campylobacter is accompanied by increased consumption of Vitamin B12 (conversion into inactive cobalamide) and by bacterial synthesis of substances that compete with the vitamin for receptors in the ileum mucosa (Othman, Crooks, and Card 2017). This further reduces the availability of Vitamin B12 (Fig. 3). In older persons (>65 years), atrophy of the gastric mucosa is frequently due to an infection with Helicobacter pylori. As many as 60% of elderly persons exhibit an increased infestation of the gastric mucosa with Helicobacter pylori and thus a high risk of developing chronic atrophic gastritis (Grober, Kisters, and Schmidt 2013). The influence of PPIs on malabsorption of vitamin B12 is documented by several studies showing a significant and dose-dependent relationship (Saltzman et al. 1994; Schenk et al. 1999; Marcuard, Albernaz, and Khazanie 1994; Bradford and Taylor 1999; Linder, Tamboue, and Clements 2017; Qorraj-Bytyqi et al. 2018). There are, however, some conflicting data in elderly patients suggesting that the PPI use for more than 12 months or even after treatment for up to 7 years does not increase the risk of vitamin B12 deficiency (Maes, Fixen, and Linnebur 2017; Schenk et al. 1996; Nehra et al. 2018). But PPIs may influence negatively the vitamin B12 status under particular risk conditions with age being one of them. In the elderly deficiencies of folate, vitamin B6 and vitamin B12 are frequent. Vitamin B12 deficiency is widespread in the elderly in particular and has been diagnosed in 10-40% of “healthy” persons above the age of 65, depending on the marker for deficiency and the cutoff used (Lindenbaum et al. 1994; Allen 2009; Obeid et al. 2004; Herrmann et al. 2003). In a case-control study among patients aged 65 years or older with documented serum vitamin B(12) status chronic (12 month) or current use of PPIs was associated with a significantly increased risk for vitamin B12 deficiency (OR 4.45; 95% CI 1.47-13.34) even after adjusting for multiple factors (age, gender, multivitamin use, or Helicobacter pylori infection). No association was found between past or short-term current use of PPI and vitamin B12 deficiency (Valuck and Ruscin 2004). Another cross-sectional study in 659 adults, aged 60-102 years from long-term care facilities and community ambulatory care confirms a significant reduction of vitamin B12 serum levels by PPIs depending on the duration of treatment (—5.4pg/mL per month of PPI use) (Dharmarajan et al. 2008). A chronic Helicobacter pylori infection might further increase the risk for vitamin B12 deficiency. Helicobacter pylori is one of the most common causes of peptic ulcer disease worldwide and a major cause of chronic superficial gastritis leading to atrophy of gastric glands. The Helicobacter pylori related hypochlorydria can be further enhanced by a long-term treatment with PPI. A prospective cohort study with Helicobacter pylori-infected patients shows that 33% of the patients treated with omeprazole for 5 years developed an atrophic gastritis with a significant decline of serum vitamin B12 levels (T0: 340 T1: 280pmol/L, p < 0.01) (Schenk et al. 1999; Kaptan et al. 2000; Labenz et al. 1996). Furthermore, patients with a genetic polymorphism of the cytochrome P450 isozyme CYP2C19, which catalyzes the metabolism of omeprazole, are also at risk for developing a vitamin B12 deficiency. CYP2C19 polymorphism significantly affected serum vitamin B12 levels in patients on long-term therapy (>1year) with omeprazole. Genotyping of CYP2C19 may be useful for patients in need of long-term treatment with omeprazole or other PPIs (Sagar et al. 1999). Finally, a recent population- based study compared 25,956 patients having incident diagnoses of vitamin B12 deficiency between January 1997 and June 2011 with 184,199 patients without B12 deficiency. Previous and current PPIs use was significantly associated with the presence of vitamin B12 deficiency, respectively a two or more years treatment with PPIs was associated with a 65% increased risk for vitamin B12 deficiency (OR, 1.65 [95% CI, 1.58-1.73]) (Lam et al. 2013).
Diagnostic of vitamin B12 deficiency
There is no “gold standard” for laboratory chemistry confirmation of a clinically relevant Vitamin B12 deficiency. In practice, diagnosis of a Vitamin B12 deficiency is primarily done by determining the serum Vitamin B12 level (serum cobalamin level). This is a low-cost test with limited specificity and sensitivity, particularly in persons with Vitamin B12 concentrations under 450ng/L (<330pmol/L). The normal serum levels based on modern laboratory chemistry methods are 200-1000 ng/L. Levels <200ng/L (<150pmol/L) are sure signs of a B12 deficiency; but a functional B12 deficiency may also be present at levels <450 ng/L Methylmalonic acid (MMA) is considered a highly sensitive functional indicator for a Vitamin B12 deficiency. MMA is a metabolic product, the breakdown of which requires Vitamin B12 . A lack of Vitamin B12 allows MMA levels to increase significantly. This also makes it possible to detect a functional B12 deficiency in which raised MMA levels and clinical symptoms of a B12 deficiency in the form of neurological and/or haematological disorders may occur despite normal serum cobalamin levels. The normal range for serum MMA is 50-300 nmol/L. MMA determination is a highly sensitive test that can confirm a diagnosis even in the early stages of a B12 deficiency and is also a suitable indicator for treatment response ( B12 substitution).
Another indirect functional parameter of vitamin B12 status is homocysteine. This potentially toxic amino acid results from demethylation of the essential amino acid methionine, which reaction requires vitamin B12 . A deficiency of Vitamin B12 leads to accumulation and thus raised blood levels of homocysteine. A serum homocysteine level >10 pmol/L thus indicates a possible deficiency of vitamin B12 . However, homocysteine is not a specific B12 deficiency marker, since deficiencies of Vitamin B6 and folic acid can also raise the homocysteine level. Determination of methylmalonic acid and homocysteine are particularly recommended in cases of diagnostically unclarified B12 deficiency. A Vitamin B12 deficiency can be excluded with something approaching 100% certainty if levels of these metabolites are within the normal ranges (Grober, Kisters, and Schmidt 2013; Dali-Youcef and Andres 2009; Morris et al. 2002).
Recommendation for clinical practice: In summary the present data show that proton pump inhibitors (PPIs) may influence the absorption of vitamin B 12 and be associated with an increased risk for B12 deficiency. This interference should receive more attention in the future in medical practice. In particular older persons (>60 years), patients on long-treatment with PPIs (>2 years), polypharmacotherapy (e.g. diuretics, metformin) and those with atrophic gastritis, Helicobacter pylori infection and/or any cause of hypochlo- rhydria should be screened at least once a year for vitamin B12 deficiency (e.g. serum vitamin B12 , methylmalonic acid, homocysteine). Vitamin B12 in a dosage of 250-1000 mg/ day, given orally for 1 month may be an effective treatment for B12 deficiencies not related to pernicious anemia (Grober, Kisters, and Schmidt 2013; Lindenbaum et al. 1990; Savage et al. 1994; Troxler, Hersberger, and Baumgartner 2008; Morris et al. 2002; Smith, Warren, and Refsum 2018; Andres et al. 2003; Vidal-Alaball et al. 2005; Wang et al. 2018).
PPIs and Iron
Iron is an essential trace element for almost all living organisms as it participates in a wide variety of metabolic processes including deoxyribonucleic acid (DNA) synthesis. The majority of functional Iron within the body is present in heme proteins, such as hemoglobin, myoglobin and cytochromes, which are involved in oxygen transport and/or mitochondrial electron transfer. However, as Iron can form free radicals, its concentration in body tissues must be tightly regulated because in excessive amounts, it can lead to tissue damage. Disorders of Iron metabolism are among the most common diseases of humans and encompass a broad spectrum of diseases with diverse clinical manifestations, ranging from anemia to Iron overload, and possibly to neu- rodegenerative diseases (Grober 2009; Nazanin Abbaspour, Hurrell, and Kelishadi 2014).
The physical state of Iron entering the duodenum greatly influences its absorption. At physiological pH, ferrous Iron (Fe+2) is rapidly oxidized to the insoluble ferric (Fe+3) form. Gastric acid lowers the pH in the proximal duodenum reducing Fe+3 in the intestinal lumen by ferric reductases, thus allowing the subsequent transport of Fe+2 across the apical membrane of enterocytes. This enhances the solubility and uptake of ferric Iron . When gastric acid production is impaired (for instance by PPIs), Iron absorption is reduced substantially. Dietary Iron is present in food in two forms: as either nonheme (~ 60%) or heme Iron (~ 30%). The physical state of Iron entering the duodenum greatly influences its absorption. At physiological pH, ferrous Iron (Fe+2) is rapidly oxidized to the insoluble ferric (Fe+3) form. Gastric acid lowers the pH in the proximal duodenum reducing Fe+3 in the intestinal lumen by ferric reductases, thus allowing the subsequent transport of Fe+2 across the apical membrane of enterocytes. This enhances the solubility and uptake of ferric Iron . When gastric acid production is impaired (for instance by PPIs such as omeprazole), Iron absorption is reduced (Schade, Cohen, and Conrad 1968; Ito and Jensen 2010; Betesh et al. 2015).
Proton pump inhibitors (PPIs) suppress gastric acid production, which can reduce not only the absorption of vitamin B12 but also of Iron . Since gastric acid plays an important role in the absorption process of Iron the longterm use of PPIs, which suppress the production of gastric acid, has been shown to increase the risk of Iron deficiency (Ito and Jensen 2010). Gastritis-induced achlorhydria can be an independent cause of Iron deficiency anemia (Betesh et al. 2015). A retrospective cohort study showed all hematologic indices from baseline, including hemoglobin, hematocrit and mean corpuscular volume significantly decreased after chronic PPI use (at least for 1year) (P < 0.01) compared to matched controls (Sarzynski et al. 2011). In a large community-based case-control study among patients without known risk factors (e.g. endurance athletes, infants, frequent blood donors, vegetarians, vegans, women, any cause of hypochlorhydria, von Willebrand disease) for Iron deficiency, long-term PPI use for >2 years was associated with an increased subsequent risk of Iron deficiency (adjusted odds ratio, 2.49; 95% confidence interval, 2.35-2.64). The risk increased with increasing potency of acid inhibition and decreased after medication discontinuation (P-trend <0.001) (Lam et al. 2017). Case reports of patients that developed Iron malabsorption because of taking PPI for years underline the problem between long-term PPI use and the potential side effect of Iron deficiency anemia (Dado, Loesch, and Jaganathan 2017; Imai et al. 2018). Furthermore, several studies indicate that many patients with unexplained Iron deficiency anemia suffer from autoimmune gastritis, and about 50% of them have evidence of a Helicobacter pylori infection. Interestingly, in patients with unexplained Iron deficiency anemia and Helicobacter pylori infection, cure of refractory Iron deficiency anemia by Helicobacter pylori eradication offers strong evidence for a cause and effect relationship between Helicobacter pylori infection and the development of Iron deficiency anemia (Valiyaveettil et al. 2005; Chen and Luo 2007). Thus, Helicobacter pylori infection may be regarded as a risk factor for reduction in body Iron stores and also for Iron deficiency or Iron deficiency anemia, especially in high-risk groups. The results of a meta-analysis revealed an increased risk for Iron deficiency anemia (OR: 2.8 (95%, CI: 1.9, 4.2) and also for Iron deficiency (OR 1.38 (95% CI: 1.16-1.65) among Helicobacter pylori-infected patients. The biologic mechanisms by which Helicobacter pylori induces the disorder in the Iron status is not fully understood, but it seems that several pathways are involved: decrease in the absorption of dietary Iron , gastrointestinal blood loss, and enhanced uptake of Iron by the bacterium (Muhsen and Cohen 2008; Hershko and Ronson 2009; Flores et al. 2017).
Diagnostic of Iron deficiency
To detect an Iron deficiency serum ferritin <30 mg/L is a sensitive and specific test to identify isolated Iron deficiency, as it reflects low Iron stores. In the progression of the Iron deficiency, because of low Iron and increased transferrin synthesis, transferrin saturation (or the saturated Iron binding capacity) drops <16%, di-ferric transferrin, the ligand of transferrin receptor, is reduced and Iron supply to the bone marrow becomes insufficient. At this point serum ferritin is usually <13 mg/L. Low transferrin saturation is one criterion with ferritin levels empirically set at <100 mg/L or higher (<300 mg/L) as in chronic kidney disease or in heart failure. Other laboratory tests for assessment of Iron status, such as the levels of serum soluble transferrin receptor (increased in Iron deficiency and normal/low in inflammation) or the ratio between soluble transferrin receptor and log ferritin levels, can also be used in clinical practice (Worwood 1997; Walsh et al. 2011; Thomas et al. 2013; Camaschella 2015).
Recommendation for clinical practice: Although there are currently no recommendations regarding screening for Iron deficiency and/or anemia in patients on long-term PPI therapy, physicians should be aware of this potential side effect and consider monitoring in high-risk patients (e.g. age >60, Helicobacter infection, any cause of hypochlorhydria, frequent blood donors, vegetarians, vegans).
PPIs and Magnesium
Magnesium is primarily found within the cell where it acts as a counter ion for the energy-rich ATP and nuclear acids. Magnesium is a cofactor in more than 600 enzyme systems that regulate diverse biochemical reactions in the body, including protein synthesis, muscle and nerve transmission, neuromuscular conduction, blood glucose control, and blood pressure regulation. Some Magnesium dependent enzymes are Na+/K+-ATPase, hexokinase, creatine kinase, protein kinase, and cyclases. Magnesium is also necessary for structural function of proteins, nucleic acids or mitochondria. It is required for DNA and RNA synthesis, reproduction, and for both aerobic and anaerobic energy production - oxidative phosphorylation and glycolysis - either indirectly as a part of Magnesium -ATP complex, or directly as an enzyme activator.
Magnesium also plays a key role in the active transport of calcium and potassium ions across cell membranes, a process that is important to nerve impulse conduction, muscle contraction, vasomotor tone and normal heart rhythm. Hypomagnesaemia is frequently linked with hypokalemia owing to disturbances in renal K + secretion in the connecting tubule and collecting duct. Magnesium is a natural calcium antagonist - the block of NMDA receptor channels by external Magnesium is believed to be of great physiological importance. Moreover it contributes to the structural development of bone and is required for the adenosine triphosphate-dependent synthesis of the most important intracellular antioxidant glutathione. Magnesium absorption and excretion is influenced by different hormones. It has been shown that 1,25-dihydroxy vitamin D [1,25(OH)2D] can stimulate intestinal Magnesium absorption. On the other hand Magnesium is a cofactor that is required for the binding of vitamin D to its transport protein, vitamin D binding protein (VDBP). Moreover, conversion of vitamin D by hepatic hydroxylation and renal 1a- hydroxylation into the active, hormonal form 1,25(OH)2D is Magnesium -dependent. Magnesium deficiency, which leads to reduced 1,25(OH)2D and impaired parathyroid hormone response, has been implicated in ‘ Magnesium -dependent vitamin-D-resistant rickets’. Magnesium supplementation substantially reversed the resistance to vitamin D treatment (de Baaij, Hoenderop, and Bindels 2015; Grober, Reichrath, and Holick 2015).
Of special importance is parathyroid hormone (PTH). Absorption of both Magnesium and calcium appears to be inter-related, with concomitant deficiencies of both ions well described. A common link is that of PTH, secretion of which is enhanced in hypocalcemia. Hypomagnesemia impairs hypocalcaemic-induced PTH release, which is corrected within in minutes after infusion of Magnesium . The rapidity of correction of PTH concentrations suggests that the mechanism of action of Magnesium is enhanced release of PTH. PTH release enhances Magnesium reabsorption in the kidney, absorption in the gut and release from the bone (Grober, Reichrath, and Holick 2015; Anast et al. 1976; Zofkova and Kancheva 1995).
Long-term use of PPIs has been associated in some cases with hypomagnesemia, hypocalcemia and hypoparathyroidism. Since the year 2006 there have been more than 40 reported cases of PPI-induced hypomagnesaemia (<0.76mmol/l). Thus in 2011, the US Food and Drug Administration (FDA) published a safety announcement, including hypomagnesaemia as a long-term side-effect of PPI based on accumulating evidence (Epstein, McGrath, and Law 2006; Luk et al. 2013; Tamura et al. 2012). A severe Magnesium deficiency with PPI use can occur in rare cases and accounts for less than 1% of all PPI-induced side-effects voluntarily reported to the FDA. Others reported hypomag- nesaemia in 13% of PPI users. Nevertheless, patients on long-term treatment with PPIs should be monitored for Magnesium deficiency, especially those with diabetes and cardiovascular disease (e.g. arrhythmias, heart failure, hypertension), because the probability and the risk of disease- associated complications is increased (Groober, Reichrath, and Holick 2015; Hess et al. 2017; Lemon 2013).
The mechanisms by which PPIs can interfere with intestinal Magnesium absorption is still being investigated. PPIs such as omeprazole may decrease intestinal Magnesium absorption by interfering with both passive (paracellular pores) absorption and active (transient receptor potential melastatin protein channels, TRPM) absorption. Common single nucleotide polymorphisms in the gene TRPM6 have been identified and are discussed to be responsible for the risk of hypomagnesaemia in PPI treated patients (Lemon 2013).
Magnesium deficiency and hypomagnesemia (<0.76 mmol/L) are a relatively common in clinical practice. There are often unrecognized because of the fact that Magnesium status is rarely controlled since only a few clinicians are aware of the many clinical states in which Magnesium deficiency can manifest (Henzel, DeWeese, and Ridenhour 1967). Despite normal serum Magnesium levels a Magnesium deficiency can be present (Rude et al. 1998). In older publications the prevalence of marginal Magnesium deficiency in developed countries is estimated to be 15-20%. Recent population-based cross-sectional studies and clinical trials indicate that some 10-30% of a given population, considered healthy, have a subclinical Magnesium deficiency based on serum Magnesium levels <0.80mmol/L (Durlach 1989; Costello et al. 2016). In postmenopausal women with osteoporosis Magnesium deficiency has been found in 84% diagnosed by low Magnesium trabecular bone content and Thoren’s Magnesium load test (Cohen and Kitzes 1981). In an ageing population, the number of patients treated with diuretics is increasing, as is the significance of diuretic therapy-associated side effects (Touitou et al., 1987). In the elderly the prevalence of Magnesium and potassium deficiencies is about 20% (Malon et al. 2004). The concentration of intracellular ionized Magnesium is physiologically relevant. Thus ionized Magnesium in erythrocytes is one of the best laboratory parameters to judge Magnesium deficiency. Among critically ill postoperative patients, 36.5% were found to have Magnesium deficiency based on ionized Magnesium levels in red blood cells (Malon et al. 2004). Based on a reference range for serum Magnesium <0.76 mmol Mg/L the frequency of hypomagnesemia was evaluated in an unselected population group of about 16,000 individuals of Germany. Hypomagnesemia was present in about 14.5% of all individuals with generally higher frequencies in females and outpatients. In the elderly, especially in old ladies, the prevalence was highest and concerned about 30% of this subgroup. In general, in this study suboptimal Magnesium levels were observed in 33.7% of the population (Schimatschek and Rempis 2001; Vormann 2003).
Hypomagnesemia can cause serious neuromuscular and cardiovascular problems and is often accompanied by hypocalcemia, hypovitaminosis D and hypokalemia. Early signs of Magnesium deficiency are non-specific and include loss of appetite, lethargy, nausea, vomiting, fatigue, muscle cramps, weakness and lethargy. More pronounced Magnesium deficiency presents with symptoms of increased neuromuscular irritability such as tremor of the extremities, tetany, generalized seizures or convulsions. Hypomagnesemia can cause hypokalemia with electrocardiogram changes and cardiac arrhythmias including atrial and ventricular tachycardia, prolonged QT interval and torsades de pointes. Carpopedal spasms are often associated with hypoparathyroidism, hypovitaminosis D and/or hypocalcemia. Additionally, the neuromuscular symptoms of hypomagnesaemia (e.g. convulsions, muscle weakness, tetany) can also be related to the co-existent hypovitaminosis D (25(OH)D: <20ng/mL) and/or hypocalcemia (Grober, Reichrath, and Holick 2015; Florentin and Elisaf 2012; Famularo, Gasbarrone, and Minisola 2013).
Diagnostic of Magnesium deficiency
The most common and valuable test in clinical medicine for the rapid assessment of changes in Magnesium status is the serum Magnesium concentration, even though serum levels have little correlation with total body Magnesium levels or concentrations in specific tissues . In healthy individuals, Magnesium serum concentration is closely maintained within the physiological range. The normal reference range for the Magnesium in blood serum is 0.76-1.05 mmol/L. A serum Magnesium <0.82mmol/L (2.0mg/dL) with a 24- hour urinary Magnesium excretion of 40-80 mg per day is highly suggestive of Magnesium deficiency. According to many Magnesium researchers the appropriate lower reference limit of the serum Magnesium concentration should be 0.85 mmol/L, especially for patients with diabetes. Furthermore, the ionized Magnesium concentration and the Magnesium loading (or tolerance) test have been shown to be more accurate. The reference range for serum ionized Magnesium concentration is 0.54-0.67 mmol/L. To comprehensively evaluate Magnesium status, both laboratory tests and the clinical assessment of Magnesium deficit symptoms might be required (Grober, Reichrath, and Holick 2015; Rude et al. 1998; Famularo, Gasbarrone, and Minisola 2013).
Recommendations for clinical practice: Patients on long-term treatment with PPIs should be monitored for Magnesium deficiency, particularly those with additive risk factors, such as therapy with use of diuretics, diabetes, cardiovascular diseases (e.g. hypertension, arrhythmias), inadequate dietary intake, secondary aldosteronism and kidney dysfunction. Many nutritional experts feel the ideal intake for Magnesium should be based on the body weight (e.g. 4-6 mg per kg/d). In the treatment of Magnesium deficiency supplements with organic bound Magnesium salts are recommended, such as Magnesium citrate or gluconate. But also mineral water with a high concentration of Magnesium (>100 mg Mg/L) is a good source of Magnesium that contributes to daily Magnesium supply (Groober, Reichrath, and Holick 2015; Schneider, Greupner, and Hahn 2017).
Thiazide diuretics
Thiazide-type diuretics are the second most commonly prescribed class of antihypertensive medication, and thiazide- related diuretics have increased at a rate greater than that of antihypertensive medications as a whole. For more than 5 decades thiazide diuretics (TD), including thiazide-type (e.g. hydrochlorothiazide chlorothiazide) and thiazide-like diuretics (e.g. indapamide, chlorthalidone) have been used for the treatment of hypertension. The latest hypertension guidelines have underscored the importance of thiazide diuretics for all patients, but particularly for those with salt-sensitive and resistant hypertension (Roush and Sica 2016). Thiazide-type diuretics decrease efficaciously systolic and diastolic blood pressure and reduce at the same time cardiovascular morbidity and mortality associated with hypertension. A meta-analysis including 19 randomized controlled trials enrolling 112,113 patients showed that thiazide diuretics have an additional cardioprotective effect. During a mean follow up of 3.91-years, a 14% reduction in the risk of cardiac events (odds ratio (OR): 0.86, P = 0.007) and 38% reduction in the risk of heart failure (OR: 0.62, P < 0.001), were found in thiazide treated patients (Chen et al. 2015). Another recent systematic review of the Cochrane Hypertension Group of 24 randomly assigned trials with 58,040 hypertensive patients (mean age ±62 years) shows with high-quality evidence that first-line low-dose thiazides reduced mortality (11.0% with control versus 9.8% with treatment; RR 0.89, 95% CI 0.82-0.97); total CVS (12.9% with control versus 9.0% with treatment; RR 0.70, 95% CI 0.64-0.76), stroke (6.2% with control versus 4.2% with treatment; RR 0.68, 95% CI 0.60-0.77), and coronary heart disease (3.9% with control versus 2.8% with treatment; RR 0.72, 95% CI 0.61-0.84) (Wright, Musini, and Gill 2018). Additionally, thiazide and loop diuretics are important tools in the therapy of volume-overload conditions, such as congestive heart failure, nephrotic syndrome, and cirrhosis, by improving the symptoms of fluid congestion, volume overload and edema. Although thiazide-type diuretics are among the best tolerated antihypertensive drugs they are often associated related adverse side effects, such as electrolyte, acid- base and/or metabolic disorders (e.g. impaired glucose tolerance, dyslipidemia) (Shen et al. 2013). All type of diuretics promote excretion of sodium. Depending upon the site and mode of action, some diuretics increase excretion of potassium, Magnesium , chloride, calcium, or bicarbonate (Fig. 4). In general electrolyte disorders, such as hyponatraemia, hypo-/hyperkalaemia are well considered and monitored in clinical practice therefore they are not further discussed at this point. For instance, hyperkalaemia due to potassium- increasing drug-drug interactions is a clinically important, but well described adverse drug event (Eschmann et al. 2014; Martin-Perez et al. 2016). Instead, in order to provide a review of current knowledge, we will focus in previously more neglected drug-nutrient interactions between thiazide- type diuretics and Magnesium .
Thiazide diuretics and Magnesium
In general, thiazide diuretics are associated with a decrease of serum Magnesium levels by 5-10%. Whereby the drug- induced Magnesium depletion is be more severe in the elderly. Up to 50% of treated patients have cellular Magnesium depletion, regardless of normal serum concentrations. Hypomagnesemia occurs more often in the elderly, and in those receiving continuous high-those diuretic therapy which may increase cardiovascular morbidity and mortality (Martin and Milligan 1987; Kroenke, Wood, and Hanley 1987). About 80% of hypertensive patients treated for at least 6 months with hydrochlorothiazide have been found to have Magnesium depletion based on retention of a parenterally administered Magnesium load, even though their Magnesium serum levels were normal (Seelig, 1990). In an elderly population of a Somerset village 48% of the thiazide- treated patients were hypomagnesaemic and 28% of the thiazide-treated patients were hypokalaemic. Thus, Magnesium and potassium depletion are commonly associated with thiazide therapy in the elderly (Petri et al. 1986; Arampatzis et al. 2013). Hypomagnesemia is often associated with hypokalemia, hypocalcemia, hypophosphatemia and hyponatremia (Whang et al. 1984). Hypokalemia, hypocalcemia and/ or hypovitaminosis D found in association with low serum Magnesium blood levels can prove refractory to all treatment measures until the underlying Magnesium deficiency is corrected (Grober, Reichrath, and Holick 2015; Florentin and Elisaf 2012; Famularo, Gasbarrone, and Minisola 2013; Dyckner and Wester 1981). Remarkable are the results of a cross-sectional study in hypertensive patients that determined serum and mononuclear cell Magnesium concentrations. This study shows that although the patients had normal serum Magnesium , thiazide diuretics can induce intracellular Magnesium depletion not detectable by assessment of blood serum (Malini et al. 1990). Therefore the serum Magnesium level reflects only a small part of total body content Magnesium . In a patient with clinical Magnesium deficiency cellular Magnesium concentration can be low despite normal Magnesium levels in blood serum (Reinhart 1988).
Furthermore it has been shown that Magnesium is a kind of second messenger for insulin action. Magnesium plays a crucial role in glucose and insulin metabolism, mainly through its impact on tyrosine kinase activity of the insulin receptor, by transferring the phosphate from ATP to protein. Magnesium may also affect phosphorylase b kinase activity by releasing glucose-1-phosphate from glycogen. In addition, Magnesium may directly affect glucose transporter protein activity 4 (GLUT4), and help to regulate glucose translocation into the cell (Groober, Reichrath, and Holick 2015; Paolisso et al. 1990). Intracellular Magnesium deficiency may affect the risk of insulin resistance and alter the glucose entry into the cell. It is imaginable that the subclin- ical Magnesium deficiency and intracellular Magnesium depletion associated with thiazide treatment may interfere with the activity of the tyrosine kinase and the insulin receptor increasing the risk of insulin resistance (Grober, Reichrath, and Holick 2015; Takaya, Higashino, and Kobayashi 2004). Patients with Magnesium deficiency show a more rapid progression of glucose intolerance and have an increased risk for insulin resistance. The supplementation of Magnesium may contribute to an improvement in both islet Beta-cell response and insulin action in thiazides treated patients and in type-2 diabetics (Grober, Reichrath, and Holick 2015; Gommers et al. 2016). In a recent randomized, double-blind, clinical study with thiazide-treated hypertensive women (age: 40-65 years) the effects of Magnesium supplementation (600 mg/day) on blood pressure and vascular function were evaluated. After 6 months, the Magnesium group had a significant reduction in systolic (SBP: 144 ± 17 vs. 134±14mmHg, P = 0.036) and diastolic blood pressure (DBP: 88 ±9 vs. 81±8mmHg, P = 0.005), and as a sign of improved endothelial function a significant increase of brachial flow-mediated dilatation (FMD) (r = 0.44, P = 0.011). The constant oral supplementation of Magnesium was associated with better blood pressure control, improved endothelial function and amelioration of subclinical atherosclerosis in these thiazide-treated hypertensive women (Cunha et al. 2017; Kisters and Grober 2013).
Recommendation for clinical practice: Subclinical Magnesium deficiency is a principal driver of cardiovascular diseases such as arrhythmias, arterial calcifications, atherosclerosis, heart failure, hypertension, and/or thrombosis. In other words disorders of Magnesium metabolism are a principal, under-recognized, driver of cardiovascular disease in medical practice everyday life. Hypertensive patients on long-term treatment with thiazide diuretics should therefore be monitored for Magnesium deficiency, particularly those with additive risk factors, such as age >60, hydrochlorothiazide doses >25mg/day, insulin resistance, cardiovascular diseases (e.g. hypertension, arrhythmias), inadequate dietary intake, secondary aldosteronism and kidney dysfunction. Dietary and supplement recommendations for Magnesium please see page 7.
Statins (HMG-CoA-reductase inhibitors)
In the world statins constitute one of the most important sectors of the pharmaceutical industry and belong to the most widely prescribed drugs. These lipid lowering drugs, also known as HMG-CoA-reductase inhibitors, form the cornerstone for prevention and treatment of cardiovascular disease. A number of large-scale trials have demonstrated that statins effectively reduce cardiovascular morbidity and mortality in both primary and secondary prevention, in all age groups and independent of gender. Statins have also been shown to slow the progression or even promote regression of coronary atherosclerosis (Weng et al. 2010; Catapano et al. 2016).
Statins reduce the synthesis of cholesterol in the liver by competitively inhibiting HMG-CoA reductase activity. The reduction in intracellular cholesterol concentration induces an increased expression of low-density lipoprotein receptors (LDL-Rs) on the surface of the hepatocytes, which results in increased uptake of low-density lipoprotein cholesterol (LDL-C) from the blood and a decreased plasma concentration of LDL-C and other apoB-containing lipoproteins, including triglycerides (TG)-rich particles. The degree of LDL-C reduction is dose dependent and varies between the different statins (e.g. rosuvastatin, fluvastatin) (Weng et al. 2010). There is considerable interindividual variation in LDL-C reduction with the same dose of drug. Poor response to statin treatment in clinical studies is to some extent caused by poor compliance, but may also be explained by a genetic background involving variations in genes of both cholesterol metabolism and of statin uptake and metabolism in the liver (SEARCH Collaborative Group 2008; Chasman et al. 2012; Stroes et al. 2015).
Statins can cause adverse side-effects, including musculoskeletal complaints, known as myositis, fatigue, gastro-intes- tinal discomfort, elevation of liver enzymes, neurocognitive symptoms, peripheral neuropathy and insomnia. In addition, randomized trials have shown a small increase in the risk of incident diabetes. Statin-associated muscle symptoms (SAMS), are reported by 10% to 29% of patients in clinical practice and belong to the most prevalent side effect. They are the principal reason for statin non-adherence and/or discontinuation, contributing to adverse cardiovascular outcomes. SAMS may vary in severity from simple myalgias to severe rhabdomyolisis (Weng et al. 2010; Stroes et al. 2015; Apostolopoulou, Corsini, and Roden 2015). vitamin D insufficiency, statin-induced coenzyme Q10 depletion and/or the negative effect of statins on selenoprotein synthesis have been discussed to be implicated in the pathogenesis of myopathic symptoms.
Statins and vitamin D
vitamin D plays a crucial role in human health, while the prevalence of vitamin D deficiency worldwide is high. According to recent studies, a vitamin D deficiency is likely to be an important etiological factor in the pathogenesis of many chronic diseases. These include autoimmune diseases (e.g., multiple sclerosis, type 1 diabetes) inflammatory bowel disease (e.g., Crohn disease), infections (such as infections of the upper respiratory tract), immune deficiency, cardiovascular diseases (e.g., hypertension, heart failure, sudden cardiac death), cancer (e.g., colon cancer, breast cancer, non- Hodgkin’s lymphoma) and neurocognitive disorders (e.g., Alzheimer disease). vitamin D in its hormonally active form, 1a,25-dihydroxy vitamin D [1a,25(OH)2D; calcitriol] is not only a regulator of calcium and phosphate homeostasis, but has numerous extra-skeletal effects. 1a,25(OH)2D manifests its diverse biological effects (endocrine, autocrine, paracrine) by binding to the vitamin D receptor (VDR) found in most body cells. vitamin D receptors have been found in over 35 target tissues that are not involved in bone
metabolism. These include endothelial cells, islet cells of the pancreas, hematopoietic cells, cardiac and skeletal muscle cells, monocytes, neurons, placental cells and T-lymphocytes. It is estimated that VDR activation may regulate directly and/or indirectly a very large number of genes (0.5-5% of the total human genome i.e., up to 2000 genes). The fact that the vitamin D receptor is expressed by many tissues results in the pronounced pleiotropic effect of vitamin D hormone. 25(OH)D is the vitamin D metabolite that is measured to assess a patient’s vitamin D status. vitamin D deficiency is diagnosed when 25(OH)D < 20 ng/mL, vitamin D insufficiency is defined as 25(OH)D of 21-29 ng/mL, and 25(OH)D > 30 ng/mL is considered sufficient, with 40-60 ng/mL being the preferred range. vitamin D intoxication usually does not occur until 25(OH)D > 150 ng/mL. vitamin D intoxication is only to be expected at levels of 25(OH)D > 150 ng/mL (Holick 2007; Grdber, Kisters, and Schmidt 2013; Wacker and Holick 2013; Grober, Reichrath, and Holick 2015; Baggerly et al. 2015; Grober et al. 2016).
The physiological role of vitamin D in muscular function has been extensively studied (Holick 2009; Ceglia et al. 2010; Bischoff-Ferrari et al. 2010; Hamilton 2010). Several studies have shown a potential role of vitamin D for prevention and treatment of statin-associated muscle symptoms (SAMS). The prevalence of myalgia in statin-treated patients vitamin D insufficiency is significantly higher compared with those that not present any signs of SAMS. In a trial with 82 vitamin-D-deficient, myalgic patients, under statin therapy, 38 were given vitamin D (50,000 units/weekfor 12 weeks), with a resultant increase in serum 25(OH)D from 20.4 +/27.3 to 48.2 +/217.9 ng/mL (P < 0.0001) and resolution of myalgia in 35 (92%) (Ahmed et al. 2009; Glueck et al. 2011). The results of a retrospective cohort study and a meta-analysis of 7 studies with 2420 patients provide evidence that low vitamin D levels are associated with myalgia in statin treated patients (Palamaner Subash Shantha et al. 2014; Michalska-Kasiczak et al. 2015). A recent study with 74 men and 72 women (age 59 ±14 years) reported that statin intolerance because of myalgia, myositis, myopathy, or myonecrosis associated with low serum vitamin D can be safely resolved by vitamin D supplementation (50,000-100,000 units /week) in most cases (88-95%) (Khayznikov et al. 2015; Jetty et al. 2016). vitamin D insufficiency can lead to the development of nonspecific muscle pain and SAMS (Pereda and Nishishinya 2016; Kurnik et al. 2012). In older adults vitamin D supplementation even seems to improve the adherence to and persistence with long-term statin treatment (Wu et al. 2018). But the association between low vitamin D levels and statin-associated muscle symptoms is still conflicting. Some retrospective cohort studies do not support an association between low 25(OH)D Levels and statin- induced myalgia (Glueck et al. 2017; Iqbal et al. 2018; Thompson 2016). Given the major clinical importance of SAMS, the results of a large double-blind, placebo-controlled crossover study are needed to elucidate the validity of vitamin D supplementation in ameliorating muscle symptoms on statin therapy (Jacobson et al. 2018).
Recommendation for clinical practice: Although previous results of vitamin D treatment of SAMS from randomized trials have produced mixed results, vitamin D deficiency is common in statin users and evident in European population at prevalence rates that are concerning. Therefore vitamin D status [25(OH)D, ng/mL) should be monitored in all statin-treated patients and compensated by adequate vitamin D supplementation [e.g., 40-60IU vitamin D per kg body weight per day, 25(OH)D target value: 40-60 ng/mL or 100-150 nmol/L]. This applies, in particular to patients with cardiovascular diseases, diabetes, the elderly (>60 years) with poor nutritional status, and statin treated patients with muscular disorders (Cashman et al. 2016).
Statins and coenzyme Q10
Ubiquinone, better known as Coenzyme Q10, is a vitaminlike substance present in nearly all human tissues. Coenzyme Q10 (CoQ10) is a crucial component of the oxidative phosphorylation and energy production in mitochondria, where it acts as both an electron carrier and proton translocator during cellular respiration and ATP production. It is also a structural component in mitochondrial complexes I, II and III and is essential in the stabilization of complex III. The reduced form of CoQ10, ubiquinol, acts as an antioxidant in both mitochondria and lipid membranes by either scavenging free radicals directly or in conjunction with vitamin E and/or vitamin C. Furthermore, it plays a pivotal role in redox control of cell signaling, stimulation of gene expression, regulation of cell growth, inhibition of apoptosis, control of thiol groups, production of hydrogen peroxide and control of membrane channels. Low CoQ10 levels are associated with complex mitochondrial disorders, driving the process of ageing and are important in the development of age-associated disease (e.g. inflammation, diabetes, cardiovascular, neurodegenerative diseases) (Frei, Kim, and Ames 1990; Ernster and Forsmark-Andree 1993; Crane 2001; Hermndez-Camacho et al. 2018; Gutierrez- Mariscal et al. 2018).
The mevalonate pathway is one of the most important metabolic processes within the cell regulated by enzyme 3- hydroxy-3-methylglutaryl-coenzyme (HMG-CoA) reductase. The HMG-CoA-reductase converts HMG-CoA to mevalo- nate. This is further transformed into isopentenyl pyrophosphate (Isp), which serves as substrate for the synthesis of several crucial cell components, such as farnesyl pyrophosphate (Farnesyl-PP), squalene, cholesterol, coenzyme Q10 (Ubiquinone) and amongst other things the generation of selenoproteins (Fig. 5) (Stroes et al. 2015; Tricarico, Crovella, and Celsi 2015; Ramachandran and Wierzbicki 2017). HMG-CoA reductase is a key pharmacological target of the cholesterol lowering statins. Statins inhibit the production of mevalonate, a precursor of both cholesterol and ubiquinone. Ubiquinone is a crucial compound for mitochondrial function and the provision of energy for cellular processes (Granler, Ericsson, and Dallner 1994; McKenney 2003). Statin-induced depletion of ubiquinone may attenuate electron transfer in the electron transport chain with deleterious effects on muscle function (Folkers et al. 1990; Watts et al. 1993; Ghirlanda et al. 1993; Ellis and Scott, 2003; Littarru and Langsjoen 2007; Deichmann, Lavie, and Andrews 2010; Mas and Mori 2010; Michalska-Kasiczak et al. 2015; Mabuchi et al. 2005; Morrison et al. 2016). Several studies in patents with SAMS also demonstrate a significant decrease of muscle coenzyme Q10 level, but it’s not clear if this is causal to myopathic symptoms (Marcoff and Thompson, 2007; Lamperti et al. 2005). A double-blind, randomized placebo-controlled trial compared the effect of the combination of coenzyme Q10/atorvastatin to standard congestive heart failure (CHF) treatment versus therapy with atorvastatin alone. The combination of daily 10 mg atorvas- tatin and 100 mg CoQ10, as an adjunctive treatment of CHF for 4 months significantly increased ejection fraction (P = 0.006) and improved New York Heart Association (NYHA) function class (P = 0.002) in comparison with the placebo group that received atorvastatin alone (Pourmoghaddas et al. 2014). In a randomized study patients with SAMS received either daily coenzyme Q10 (100mg/day) or vitamin E (400IU/day) for 30 days. Muscle pain and pain interference with daily activities were assessed before and after treatment. After a 30 days treatment with coenzyme Q10 pain severity decreased significantly by 40% (p < 0.001) and pain interference with daily activities decreased by 38% (p < 0.02). In contrast, no changes in pain severity (+9%, p = NS) or pain interference with daily activities (—11%, p = NS) was observed in the vitamin E group (Caso et al. 2007). But recent data from randomized trials on coenzyme Q10 reducing SAMS are conflicting (Banach, Serban, Ursoniu, et al. 2015; Littlefield, Beckstrand, and Luthy 2014; Banach, Serban, Sahebkar, et al. 2015). A randomized trial examined the effect of coenzyme Q10 on simvastatin-associated muscle pain, muscle strength and aerobic performance. 41 patients with SAMS were randomized to simvastatin 20 mg/d combined with coenzyme Q10 (600 mg/d ubiquinol) or placebo for 8 weeks. Muscle pain, time to pain onset, arm and leg muscle strength, and maximal oxygen uptake were measured before and after each treatment. Pain severity and interference scores increased with simvastatin therapy, irrespective of coenzyme Q10 treatment (p = 0.53 and 0.56). There were no changes in muscle strength or VO2max with simvastatin with or without CoQ10 (all p > 0.10). Marginally more subjects reported pain with coenzyme Q10 (14 of 20 vs 7 of 18; p = 0.05). There was also no difference in time to pain onset in the coenzyme Q10 (3.0 ± 2.0 weeks) vs. placebo (2.4 ±2.1 wks) groups (p = 0.55) (Taylor et al. 2015). Another randomized trial tested whether coenzyme Q10 supplementation could decrease SAMS. After 30 days of treatment the intensity of muscle pain, measured as the Pain Severity Score (PSS), in the coenzyme Q10 group (50 mg twice daily) was significantly reduced from 3.9 ±0.4 to 2.9 ±0.4 (P < 0.001). The Pain Interference Score (PIS) also was reduced from 4.0 ±0.4 to 2.6 ±0.4 (P < 0.001). Whereas in the placebo group both parameter did not change. Coenzyme Q10 supplementation decreased statin-related muscle symptoms in 75% of patients. The relative values of PSS and PIS significantly decreased (—33.1% and —40.3%, respectively) in the coenzyme Q10 compared to placebo group (both P < 0.05). The present study shows that coenzyme Q10 effectively reduced SAMS, causing lower interference of statin-related muscular symptoms with daily activities (Skarlovnik et al. 2014).
Recommendation for clinical practice: Although previous trials of coenzyme Q10 for treatment of SAMS have produced mixed results, coenzyme Q10 appears to be safe. Supplementation of coenzyme Q10 (e.g. 100mg/day) can be an effective alternative to statin discontinuation, especially in patients with high cardiovascular and/or cerebrovascular risk (Ayers et al. 2018; Hermndez-Camacho et al. 2018; Taylor, 2018).
Statins and Selenium
Selenium is an essential micronutrient for human health whose biological activities and anti-carcinogenic properties likely result from its incorporation as the 21st proteinogenic amino acid selenocysteine in selenoproteins encoded by 25 separate human genes with roles in cell protection from oxidative stress, redox control, and the inflammatory response. The production, metabolism and action of thyroid hormone in target tissues depend on Selenium availability. Selenium - dependent glutathione peroxidases and thioredoxin reductases are necessary for optimal function of immune cells by controlling oxidative stress and redox regulation. Specific selenoproteins also have ROS-independent roles in modulating inflammatory responses. Low Selenium status has been associated with increased risk of mortality, poor immune function, cognitive decline, increased risk for autoimmune thyroid disease (e.g. Hashimoto Thyreoiditis). Prospective studies have shown some benefit of higher Selenium status on the risk of different cancers such as prostate, lung, colorectal, and bladder cancers (Rayman, 2012; Kohrle, 2015; Grober et al. 2016; Schomburg, 2016; Zhang, Zeng, and Cheng 2018).
The muscular side-effects associated with statin-treatment resemble in some ways the symptoms of Selenium deficiency. Statins can interfere with biochemical pathway of the isopentenylation of selenocysteine-tRNA (Fig. 5). The negative effect of statins on selenoprotein synthesis seems to explain some of muscular side-effects of statins, in particular, statin-induced myopathy (Moosmann and Behl, 2004). Selenoprotein N is ubiquitously expressed and essential for muscle regeneration. Selenoprotein N deficiency causes several neuromuscular disorders collectively termed as SEPN- related myopathies, characterized by early onset, generalized muscle atrophy, and muscle weakness affecting especially axial muscles and leading to spine rigidity, severe scoliosis, and respiratory insufficiency (Castets et al. 2012). In a pilot study the benefits of the combination of Selenium and coenzyme Q10 administered to patients with SAMS were evaluated. Blood levels of coenzyme Q10 increased from 0.81 ±0.39 to 3.31 ±1.72 pmol/L in the active group of patients compared with the placebo (p = 0.001). Also, the symptoms of SAMS significantly improved in the active group (p < 0.001): the intensity of muscle pain decreased from 6.7 ±1.72 to 3.2 ±2.1 (p < 0.01, —53.4 ± 28.2%); muscle weakness decreased from 7.0 ±1.63 to 2.8 ±2.34 (p < 0.01, —60 ±24.0%); muscle cramps decreased from 5.33 ±2.06 to 1.86 ±2.42, p < 0.01, —65 ±28%); tiredness decreased from the initial 6.7 ±1.34 to 1.2 ±1.32 (p < 0.01, —82 ±22%). Any significant changes could be seen in the placebo group. Notably, additional Selenium supplementation was not associated with any statistically significant decrease of SAMS (Fedacko et al. 2013). Another study evaluated also the effects of Selenium and coenzyme Q10 supplementation on SAMS. Following a 6-week washout period during which no statins were administered, the patients were re-challenged with 10 mg of atorvastatin. Patients who experienced SAMS continued the atorvastatin treatment and were in addition randomized to receive 12 weeks supplement of 400 mg Q10 and 200 mg Selenium per day or a matching double placebo. SAMS was assessed using 3 validated symptom questionnaires, and a muscle function test was performed at the beginning and at the end of the study. The patients receiving the active supplement experienced significant increases in their serum Q10 and Selenium concentrations compared with the group receiving placebo. But no statistically significant differences in symptom questionnaire scores or muscle function tests were revealed between the groups (Bogsrud et al. 2013). These results are somewhat suprising, because Selenium plays a crucial role in the metabolism of coenzyme Q10. Low cardiac contents of Selenium and coenzyme Q10 have been found in patients with cardiomyopathy. A vital relationship exists between the two substances to obtain optimal function of the cell. The selenoprotein thioredoxin reductase (TrxR) is an essential antioxidant enzyme that reduces ubiquinone-10 and thereby regenerated the antioxidant ubiquinol-10, which is important for protection against lipid and protein peroxidation (Xia et al. 2003; Alehagen and Aaseth, 2015). In a prospective randomized doubleblind placebo-controlled trial among elderly with cardiomyopathy long-term supplementation of Selenium combined with coenzyme Q10 reduced cardiovascular mortality. The positive effect could also be seen in the cardiac biomarker N-terminal proBNP (NT-proBNP) that was significantly lower in the combination group compared with the placebo group (mean values: 214ng/L vs. 302ng/L at 48 months; P = 0.014) (Alehagen et al. 2013). After 10-year and 12-year follow-up in a group of healthy elderly participants given four years of intervention with Selenium and coenzyme Q10, still a significant reduction of cardiovascular mortality was observed (Alehagen, Aaseth, and Johansson 2015; Alehagen et al. 2018). Remarkably, a subgroup analysis of this doubleblind placebo-controlled trial found significant differences in expression of more than 100 different microRNAs with up to 4 fold differences as a result of the intervention of Selenium and coenzyme Q10 combined. These changes in microRNA could explain partly the underlying mechanisms of the clinical effects of Selenium combined with coenzyme Q10, reported earlier, that reduced cardiovascular mortality, increased cardiac bioenergetics, and decreased signs of inflammation and oxidative stress (Alehagen et al. 2017). A secondary analysis of this study revealed the important impact of Selenium status on cardiovascular mortality. A reduced Selenium concentration, 67 mg/L, was found among the participants, and the cardiovascular mortality was higher in the subgroup with the lower Selenium concentrations <65 mg/L in comparison with those having a Selenium concentration >85 mg/L. Especially, the supplementation of Selenium and coenzyme Q10 was cardio-protective in those with a low Selenium concentration, <85 mg/L at inclusion. In those with serum Selenium >85 mg/L and no apparent deficiency, there was no effect of supplementation (Alehagen, Alexander, and Aaseth 2016; Alehagen et al. 2018).
Recommendation for clinical practice: The studies presented here indicate that Selenium plays an important role in coenzyme Q10 status and its multiple cardioprotective functions. Supplementation of Selenium and coenzyme Q10 can be recommended in statin and SAMS treatment, especially in patients with Selenium deficiency and high cardiovascular risk. Efforts should be made to achieve a target blood Selenium level between 130 and 150 mg/L.
Metformin
Metformin is one of the most widely used oral hypoglycemic agents. Current global clinical practice recommendations propose that metformin should be initiated with concurrent lifestyle modifications at initial diabetes diagnosis. The management of type 2 diabetes requires aggressive treatment to achieve glycemic and cardiovascular risk factors. In this setting metformin is used not only for its antihyperglycemic properties but also for its effects beyond glycemic control such as improvements in endothelial dysfunction, insulin sensitivity, oxidative stress, and lipid profiles (Rojas and Gomes 2013; Knowler et al. 2002).
Metformin and vitamin B12
Vitamin B12 deficiency is an often and insidious side effect from long-term treatment with Metformin. Its prevalence among metformin users is approximately 9.5-20%. In randomized trials serum vitamin B12 levels have been reported to be inversely associated with the dose and duration of metformin use. In a case-control study carried out on a Chinese population found that for each daily dose increment of 1000 mg/d of metformin, the odds ratio to develop vitamin B12 deficiency was 2.88 (95% CI, 2.15-3.873). Similarly, when treatment duration was longer than 3 years, the odds ratio for a vitamin B12 deficiency was 2.39 (95% CI, 1.46-3.91) (De Jager et al. 2010; Ting et al. 2006). A systematic review of six randomized controlled trials showed that serum vitamin B12 concentrations are significantly lower in patients treated with metformin (P = 0.0001) and the decrease of vitamin B12 levels is induced in a dose dependent manner, especially at daily doses >2000 mg Metformin (Liu et al. 2014). The underlying mechanism of his interaction is still discussed. Most probably, metformin may interfere with the active calcium dependent absorption of the vitamin B12 -intrinsic factor complex (Fig. 3). The concomitant intake of metformin with calcium seems to attenuate the effect on vitamin B12 absorption (Bauman et al. 2000). Several trials demonstrated a possible causal relationship between Metformin-induced vitamin B12 depletion with cognitive dysfunction, peripheral neuropathy, hematologic disturbances and increased levels of homocysteine (Andres and Federici 2007; Bell 2010; Wulffele et al. 2003; Callaghan, Hadden, and Tomkin 1980). Elevated homocysteine plasma levels are a risk factor for micro- and macrovascular complications in patients with type 2 diabetes, such as retinopathy and stroke (Sato et al. 2013). A recent systematic review and meta-analysis of randomized trials confirmed these results. Since elevated homocysteine levels are a risk factor of a series of adverse clinical outcome, supplementation of vitamin B12 and folic acid might be necessary for metformin-treated patients (Zhang et al. 2016). In patients with type 2 diabetes treated with metformin the decreased vitamin B12 blood levels can be compensated with either intramuscular application of hydroxocobalamin injections (1 mg i.m.) or sublingual application of methylcobalamin (e.g. 1 mg/day) (Parry-Strong et al. 2016). Metformin not only induces vitamin B12 depletion in serum, but also progressively increases methylmalonic acid (MMA) level in blood serum. MMA is considered a highly sensitive functional indicator for a Vitamin B12 deficiency. The increase of MMA in metformin users was associated with significant worsening of the Neuropathy Score (NPS). These results support once more that metformin-induced B12 depletion is clinically relevant. Thus, all patients with type 2 diabetes treated with Metformin, especially those with concomitant use of proton pump inhibitors, should be monitored for vitamin B12 status and/or homocysteine (Long et al. 2012; Out et al. 2018).
Recommendation for clinical practice: Patients with type 2 diabetes treated with Metformin, especially those with concomitant use of proton pump inhibitors, should be monitored for vitamin B12 deficiency and/or hyperhomocys- teinemia. In particular older persons (>60 years), patients on long-treatment with metformin, polypharmacotherapy (e.g. diuretics) should be screened at least once a year for vitamin B12 deficiency (e.g. serum vitamin B12 , methylmalonic acid, homocysteine). Vitamin B12 in a dosage of 250-1000 mg/day, given orally for 1 month may be an effective treatment for B12 deficiencies not related to pernicious anemia.
Conclusion
Given the ever-increasing number of drugs on the market and the frequency with which they are used, greater attention must be paid by physicians and pharmacists in daily medical and pharmaceutical practice focused in particular on the adverse effects of drug therapy on the micronutrient status in order to minimize the potential risk to the health of patients (Table 1) Especially high-risk patients (e.g. the elderly, patients on polypharmacotherapy) and individuals under medication with drugs such as proton pump inhibitors, diuretics and/or statins should be monitored for drug induced calciotropic hormones.