MAGNESIUM IN MAN - IMPLICATIONS FOR HEALTH AND DISEASE – review 2015
Physiol Rev 95: 1-46 , 2015 doi:10.1152/physrev.00012.2014
Jeroen H. F. de Baaij, Joost G. J. Hoenderop, and Rene J. M. Bindels
Department of Physiology, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Nijmegen, The Netherlands
Google Scholar shows 1295 publications listing this item as a reference Jan 2023
Magnesium in Obesity, Metabolic Syndrome, and Type 2 Diabetes - Jan 2021 https://doi.org/10.3390/nu13020320 FREE PDF
Magnesium in Aging, Health and Diseases - Jan 2021 https://doi.org/10.3390/nu13020463 FREE PDF
Magnesium Status and Stress: The Vicious Circle Concept Revisited Dec 2020 10.3390/nu12123672 FREE PDF
Magnesium, Oxidative Stress, Inflammation, and Cardiovascular Disease - Sept 2020 https://doi.org/10.3390/antiox9100907 FREE PDF
Magnesium: The Forgotten Electrolyte—A Review on Hypomagnesemia April 2019 https://doi.org/10.3390/medsci7040056 FREE PDF
Mg2+ Transporters in Digestive Cancers 2021 https://doi.org/10.3390/nu13010210 FREE PDF
The Role of Magnesium in Pregnancy and in Fetal Programming of Adult Diseases - Dec 2020 https://doi.org/10.1007/s12011-020-02513-0 FREE PDF
-
- 4 days longer in ICU, behind a $39 paywall
- Chapter Six – The Importance of Magnesium in the Human Body: A Systematic Literature Review - 2016
- Behind a $32 paywall
Magnesium and Kidney Health - More on the ‘Forgotten Electrolyte 2016 free PDF online
Mitochondrial Mg2+ homeostasis decides cellular energy metabolism and vulnerability to stress July 2016
Magnesium (Mg2+ is an essential ion to the human body, playing an instrumental role in supporting and sustaining health and life. As the second most abundant intracellular cation after potassium, it is involved in over 600 enzymatic reactions including energy metabolism and protein synthesis. Although Mg2 + availability has been proven to be disturbed during several clinical situations, serum Mg2+ values are not generally determined in patients. This review aims to provide an overview of the function of Mg2+ in human health and disease. In short, Mg2+ plays an important physiological role particularly in the brain, heart, and skeletal muscles. Moreover, Mg2+ supplementation has been shown to be beneficial in treatment of, among others, preeclampsia, migraine, depression, coronary artery disease, and asthma. Over the last decade, several hereditary forms of hypomagnesemia have been deciphered, including mutations in transient receptor potential melastatin type 6 (TRPM6), claudin 16, and cyclin M2 (CNNM2). Recently, mutations in Mg2+ transporter 1 (MagT1)were linked to T-cell deficiency underlining the important role of Mg2+ in cell viability. Moreover, hypomagnesemia can be the consequence of the use of certain types of drugs, such as diuretics, epidermal growth factor receptor inhibitors, calcineurin inhibitors, and proton pump inhibitors. This review provides an extensive and comprehensive overview of Mg2+ research over the last few decades, focusing on the regulation of Mg2+ homeostasis in the intestine, kidney, and bone and disturbances which may result in hypomagnesemia.
INTRODUCTION
Magnesium (Mg2+) is an essential ion for health. Mg2+ plays an important role in the physiological function of the brain, heart, and skeletal muscles. Mg2+ has anti-inflammatory properties and acts as Ca2+ antagonist. The United States Food and Nutrition Board recommends a daily intake of 420 mg for men and 320 mg for women (1). However, recent reports estimate that at least 60% of Americans do not consume the recommended daily amount of Mg2+ (281). Part of the problem stems from the soil used for agriculture, which is becoming increasingly deficient in essential minerals. Over the last 60 years the Mg2+ content in fruit and vegetables decreased by 20-30% (570). Moreover, the Western diet contains more refined grains and processed food. Estimates are that 80-90% of Mg2+ is lost during food processing. As a result, a significant number of people are Mg2+ deficient, which may comprise up to 60% of critically ill patients (84, 145). Mg2+ deficiency is commonly determined by measuring total serum Mg2+ concentrations, which ranges between 0.7 and 1.05 mM in a healthy person (323). However, serum Mg2+ values reflect only 1% of the body Mg2 + content, since most of the body’s Mg2+ is stored in bone, muscle, and soft tissues. Therefore, although serum values are within the normal range, the body can be in a severely Mg2+-depleted state. Consequently, the clinical impact of Mg2+ deficiency may be largely underestimated.
The first use of Mg2+ in human medicine can be traced back to 1697 when Dr. Nehemiah Grew identified magnesium sulfate (MgSO4) as the major ingredient of Epsom salt (195). Epsom salt was extracted from a well in Epsom, England and was used over the years to treat abdominal pain, constipation, sprains, muscle strains, hyaline membrane disease, and cerebral edema. Subsequently, Mg2 + was recognized as an element (Mg) by Joseph Black in 1755 and first isolated by Sir Humphrey Davy from magnesia [Mg3SO4O10(OH)2] and mercury in 1808 (102). The role of Mg2+ in the human body emerged once Mg2+ was described in blood plasma by Willey Glover Denis in 1920 (113). In 1926, Jehan Leroy demonstrated that Mg2+ is essential for life in mice (309). These findings were translated to humans, and the first report of Mg2+ deficiency in humans was by Arthur Hirschfelder and Victor Haury in 1934 (231). Since then, Mg2+ has been implicated in and used for treatment of a variety of diseases, including migraines, cardiovascular diseases, and diabetes. Although the importance of Mg2+ is widely acknowledged, serum Mg2+ values are not generally determined in clinical medicine. Therefore, Mg2+ is often referred to as the “forgotten” cation in human health.
This review provides an overview of the role of Mg2+ in human health and disease. Mg2+ has important cellular functions in enzymatic reactions and in the synthesis and structure of proteins and polynucleotides, which are described in section II. The important regulation of Mg2+ homeostasis is discussed in depth in section III. The role of Mg2+ in organ function and related diseases is discussed in section IV. An overview is presented of the most important diseases in which Mg2+ disturbances have been implicated or in which Mg2+ has been considered as a potential treatment. In last part of the review, special attention is awarderd to disturbances of intestinal Mg2+ uptake and renal Mg2+ excretion (sect. V). All together, this review emphasizes the importance of a controlled Mg2+ balance in the human body. Increasing the awareness and understanding of Mg2+ homeostasis may give more clinical attention to the important role of Mg2+ in health and disease.
MAGNESIUM IN CELLULAR PHYSIOLOGY
Within the periodic table of elements, Mg has the atomic number 12 and is classed as an alkaline earth element (group 2). Mg occurs in three stable isotopes: 24Mg, 25Mg, and 26Mg. 24Mgis the most common isotope (78,99%) and has a relative atomic mass of 24.305 Da, a melting point of 648.8°C, and a boiling point of 1,090°C (350). Mg2+ is highly soluble and the second most abundant cation in seawater (95). In the dissolved state, Mg2+ has two hydration shells, making its hydrated radius ~400 times larger than its dehydrated radius, larger than that of other cations like Na+, K+, and even Ca2+ (95). Consequently, Mg2+ needs to be dehydrated before passing through channels and transporters, a process that requires a lot of energy. Mg2+ is a powerful Ca2+ antagonist, despite both having similar charge and chemical properties.
Mg2+ is the second most abundant intracellular cation with typical concentrations of ~ 10-30 mM. However, since most of the intracellular Mg2+ is bound to ribosomes, polynucleotides and ATP, the concentration of freely available Mg2+ falls within the low millimolar range (0.5-1.2 mM) (133). In contrast to other abundant ions, for which cells maintain considerable transmembrane gradients, the free Mg2+ concentrations in the cell and in the extracellular fluid are comparable. Mg2+ is a versatile ion that is involved in practically every major metabolic and biochemical process within the cell. Although it extends beyond the purpose of this review to give a comprehensive overview of all biochemical reactions and structural processes involving Mg2+, the following paragraphs will highlight the most prominent cellular processes in which Mg2+ is involved.
Nucleotide Binding
Mg2+ forms an essential component of the RNA and DNA tertiary structures, as it binds the negatively charged O and N molecules within the polynucleotide chains. Polynucleotide binding is a complex biophysical process that mainly depends on the level of Mg2+ dehydration and the electrostatic potential at the binding site (for extensive review, see Ref. 347). The most studied Mg2+-RNA interaction is tRNA, where Mg2+ stabilizes the structure. The role of Mg2+ became evident in 1966, when it was shown that Mg2+ could restore denaturated tRNA molecules (319). Crystallographic structures of tRNAs from yeast identified five Mg2+-binding sites, three in the core region around the bend of the L-shaped molecule and two additional sites in the major groove of the anticodon stem (412, 556). Additionally, there may be a few dozen Mg2+ in close vicinity of tRNA molecules that may bind weakly to the exterior of the structure (347). Still, the importance of Mg2+ binding for the tRNA tertiary structure has been contested over the years. This discussion was mainly triggered by studies showing the importance of nonspecific diffuse binding of Mg2+ and other divalent and monovalent cations, questioning the specificity of the Mg2+ interactions. However, the role of Mg2+ in RNA structure extends beyond tRNAs. For instance, Mg2+ is also crucial to the interactions that stabilize the pseudoknot conformation (191), tertiary RNA structures that are present in mRNA, ribosomal RNA, transfer-messenger RNA, catalytic self-splicing RNA, and viral genomic RNA.
In DNA, Mg2+ forms hydrogen bonds with the electronegative elements (O, N) to stabilize the natural DNA conformation, referred to as B-DNA (85, 549). Moreover, Mg2 + plays a role in the secondary and tertiary structure of DNA by competing with monovalent ions (394). Mg2+ binds the minor groove of B-DNA structures, thus protecting it. In Mg2+-deficient conditions, DNA is more accessible to free oxygen radicals and more prone to oxidative stress (406). However, at higher Mg2+ concentrations, Mg2+ may covalently bind DNA, locally distorting the double helix (22). Therefore, maintaining the cellular Mg2+ concentration within the physiological range is essential for DNA stability.
Enzymatic Activity
In medical textbooks and scientific litera ture, Mg2+ is often described as a cofactor for ~300 enzymes. Theodor Gunther introduced the number 300 as a rough estimate in 1980 and this has been in use ever since (133). However, in the decades after 1980 many new Mg2+-dependent enzymes have been described, and the number 300 is, therefore, an underestimation. Currently, enzymatic databases list over 600 enzymes for which Mg2+ serves as cofactor, and an additional 200 in which Mg2+ may act as activator (32, 73). An overview of these Mg2+-dependent enzymes can be found at MetaCyc (http://www.metacyc.org; Ref. 73). Many of the enzymes that require Mg2+ as coactivator are vital for life.
Mg2+ is necessary for the proper structure and activity of DNA and RNA polymerases (56, 500). DNA polymerases have two Mg2+ binding sites, which are hypothesized to play a key role in the conformational changes in the polymerase enzyme during the catalytic reaction (56). This model was further enhanced by studies reporting that the release of one of the Mg2+ions is necessary for opening the catalytic site for new nucleotides (577). In addition, Mg2 + is an important factor in DNA repair mechanisms within the cell, including nucleotide excision repair (NER), base excision repair (BER), and mismatch repair (MMR). Mg2+ acts as cofactor for almost every enzyme involved in basically every step of NER (68). In BER, Mg2+ is elemental for the activity of endonucleases, which incise the DNA after DNA damage, and the DNA polymerases and ligases, which repair the gap (41, 473). The third repair pathway, MMR, is also affected by Mg2+ availability since several enzymes involved require Mg2+ and ATP for activity (33). Other enzymes requiring Mg2+ are topisomerases, helicases, exonucleases, protein kinases, cyclases, and large groups of ATPases, meaning that Mg2+ is an essential component of DNA replication, RNA transcription, amino acid synthesis, and protein formation. Altogether, Mg2+ is a key factor in the maintenance of genomic and genetic stability. The consequences of low Mg2+ availability on the development of cancer is discussed in section IIE1.
Mg2+ is also an important regulator of many enzymes involved in glycolysis, because it is a cofactor for adenine nucleotides. Mg-ATP is required for the activity of hexokinase, phosphofructokinase, aldolase, phosphoglycerate kinase, and pyruvate kinase (166, 550). Consequently, Mg2+ availability is of major importance for glucose metabolism, which may explain its role in diabetes mellitus type 2 (see sect. IVE1).
The role of Mg2+, however, extends far beyond DNA and protein synthesis, DNA repair, and glycolysis. Since kinases, ATPases, guanylyl cyclases, and adenylyl cyclases all depend on Mg-ATP for proper function, Mg2+ plays a role in virtually every process in the cell.
Cellular Mg2+ Handling
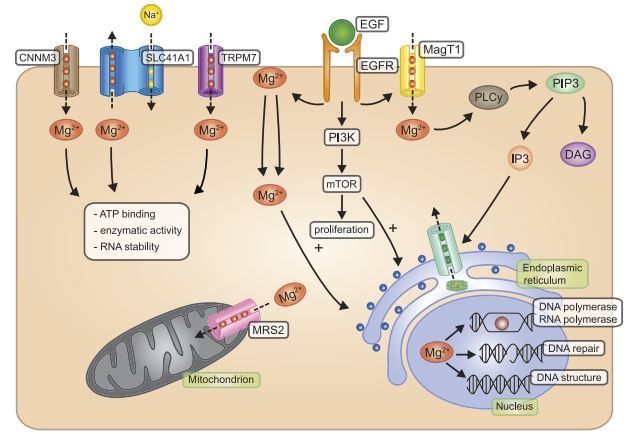
Since protein and DNA synthesis are highly dependent on intracellular Mg2+ availability, intracellular Mg2+ concentrations are tightly regulated. Over the last 20 years, elucidating the molecular identities of the transporters involved in Mg2+ homeostasis has been the main focus of research within the Mg2+ field. Genetic screenings on human diseases and microarray-based expression studies have resulted in the identification of numerous Mg2+-transporting proteins (TABLE 1 and FIGURE 1). Although the exact role of many of these proteins needs further investigation, researchers have identified several proteins critical to cellular Mg2+ homeostasis. Within this part of the review, we will focus on the ubiquitous transporters transient receptor potential melastatin type 7 (TRPM7), Mg2+ transporter 1 (MagT1), and solute carrier family 41 member 1 (SLC41A1). Tissuespecific Mg2+ transporters such as transient receptor potential melastatin type 6 (TRPM6; kidney, colon), cyclin M2 (CNNM2; kidney) and cyclin M4 (CNNM4; colon) are discussed in section III.
Figure F1
TRPM7
TRPM7 is a ubiquitously expressed divalent cation channel that is responsible for much of the Mg2+ flux in the cell. TRPM7 activity is generally regarded as a prerequisite for cell viability (360, 455). However, recent reports with tissue-specific TRPM7 KO mice suggest that TRPM7-deficient T cells are still viable and have normal intracellular Mg2+ concentrations (266). TRPM7 constitutes a tetrameric channel, where each subunit consists of six transmembrane regions with a pore region between the fifth and sixth transmembrane domain (42). The intracellular COOH terminus contains a kinase domain that regulates autophosphorylation of the channel, although its mechanism is poorly understood, as TRPM7 channel function is not dependent on its kinase activity (504). Initially, the kinase was reported to exist as separate entity (438), and indeed, recently it was shown that the kinase is cleaved from TRPM7 by caspase-8, although the exact function of the cleaved kinase remains unknown (115).
MagTI
Originally identified in MDCT cells, Mg2+ transporter 1 (MagT1) has been described as a ubiquitously expressed Mg2+ channel (187). Survival and growth of TRPM7-deficient cells can be partially rescued by MagTl overexpression (110). Although identification of MagT1 dates back almost 10 years, the functional characteristics of MagT1 are still undetermined. MagT1 mediates highly specific Mg2+ currents in Xenopus laevis oocytes, but these results could not be reproduced in mammalian cells (187, 589). Recent studies in T cells suggest that MagT1 mediates a rapidMg2+ influxuponreceptor activation (79, 311). Since T cells do not require TRPM7 for maintaining normal intracellular Mg2+ concentrations (266), this suggests that MagT1 has a similar function as TRPM7 in certain cell types.
Extrusion
In 1984, Theodor Gunther et al. (214) proposed that the main route of Mg2+ efflux from the cell is Na+ dependent. A large body of evidence obtained in a wide range of cell types supports this notion (reviewed in Ref. 424). Over the last decades, the mechanism has been further characterized in a variety of cell types, demonstrating inhibition by Na + channel blockers such as amiloride, imipramine, and quinidine. The stoichiometry of this exchange mechanism is still not fully elucidated; Na+-dependent Mg2+ extrusion is activated by cAMP in several cell models and conditions (424), but Na+-independent Mg2+ extrusion has also been proposed. Ebel et al. (134) reported the presence of a choline-dependent Mg2+ transporter in erythrocytes. However, the molecular identity of this proposed Mg2+ efflux mechanism remains controversial.
SLC41A1
Recent reports by Kolisek and co-workers (288, 289) suggested that solute carrier family 41 member 1 (SLC41A1) functions as a Na+/Mg2+ exchanger with a 2:1 stoichiometry. SLC41A1 contains 11 transmembrane domains and was originally described as a Mg2+ transporter mediating Mg2+ currents in Xenopus laevis oocytes (185). Although electrophysiological analysis could not confirm these measurements in mammalian cells, Mg2+ efflux studies using mag-fura 2 show Na+-dependent Mg2+ extrusion (251, 289). Gain-of-function SNPs have been associated with Parkinson’s disease, and one mutation in SLC41A1 was identified in a patient with a nephronophthisis-like phenotype (251, 290, 576). SLC41A1 is part of a larger protein family including two additional members, SLC41A2 and SLC41A3, which are studied less extensive. Although SLC41A2 was initially described as a plasma membrane protein, it has a topology opposite to SLC41A1 (442). This finding suggests that SLC41A2 might be expressed on the membranes of organelles and may be involved in subcellular Mg2+ transport.
CNNM3
Members of the Cyclin M (CNNM) family have been proposed to function as Mg2+ transporters (184, 545). CNNM1 is mainly expressed in brain, CNNM2 expression is high in kidney, and CNNM4 is primarily expressed in intestine (105). In contrast, CNNM3 has a ubiquitous expression pattern and may play a role in the maintenance of cellular Mg2+ homeostasis. A recent study shows that CNNM3 transports Mg2+, and its activity is regulated by oncogene PRL2 (217). The interaction between PRL2 and CNNM3 is essential for Mg2+ influx that drives tumor growth. Therefore, CNNM3 should be considered in future studies on cellular Mg2+ handling in nonpathological conditions.
MRS2
Although most studied in yeast, MRS2 (mitochondrial RNA splicing 2) is considered to be the primary Mg2+ channel on the mitochondrial membrane (592). Knockdown of MRS2 results in reduced Mg2+ uptake in mitochondria and cell death (399). Using the newly developed mitochondrial Mg2+ fluorescent probe KMG-301, Shindo et al. (475) revealed that MRS2 regulates intramitochondrial Mg2+ concentrations. This finding is interesting, since it indicates that mitochondria may store intracellular Mg2 + . Given that Mg2+ is of major importance for ATP binding, intramitochondrial Mg2+ concentrations may indirectly influence the progression of the citric acid cycle. Recently, it was shown that MRS2 mutations cause demyelination. The relevance of this observation to Mg2+ homeostasis still remains to be determined (296).
Others
In addition to the aforementioned Mg2+ transporters, several other proteins have been proposed to transport Mg2+. However, these claims are based mainly on overexpression in the Xenopus oocytes model, and functional evidence for these proteins is scarce. For example, the nonimprinted in Prader-Willi/Angelman syndrome (NIPA) family of proteins has been proposed to transport Mg2+, based on Mg2+ currents in Xenopus laevis oocytes (182), but recent studies indicate that NIPA proteins have a role in bone morphogenetic protein (BMP) signaling (525). Likewise, Huntingtininteracting protein 14 (HIP14) was thought to mediate Mg2+ fluxes at the Golgi membrane (183). Now it has become apparent that its main function consists of palmitoyl acyltransferase activity, specifically involved in the palmitoylation of Huntington (129, 582). Therefore, the role of NIPA proteins and HIP14 in Mg2+ transport should be questioned. Additionally, members of the membrane Mg2 + transporter (MMgT) family have been shown to transport divalent cations in Xenopus oocytes (186). However, as they have only one transmembrane domain after signal peptide cleavage, it is unlikely that they form functional Mg2 + transporters themselves. It is possible that MMgT proteins may form subunits of other Mg2+ channels, and as a consequence, future studies should be directed to the identification of its protein partners.
Cell Signaling
Mg2+ acts as a physiological Ca2+ antagonist within cells, and as a result, the Mg2+/Ca2+ ratio is of major importance for the activity of Ca2+-ATPases and other Ca2+ transporting proteins (257). Small changes in the Mg2+ availability within the cell may therefore cause disturbed Ca2+ signaling or Ca2+ toxicity.
Since 1974, when Mg2+ influx was detected upon insulin stimulation, several groups have suggested a second messenger role for Mg2+ (311, 322, 503). Most recently in a study on T-cell activation, MagT1 channels were shown to mediate Mg2+ influx upon T-cell receptor activation and EGF stimulation (311). In these T cells, Mg2+ activates phospholipase C-y (PLCy1), resulting in reduced phosphorylation of protein kinase C (PKC) and inositol trisphosphate (IP3)generation downstream, eventually leading to reduced Ca2+ influx (FIGURE 1). In contrast, other reports suggest that PLCy activation precedes Mg2+ influx (240, 241). The proposition of Mg2+ as a dynamic second messenger raises many questions. How do MagT1 or other Mg2+ transporters facilitate rapid Mg2+ influx when the intracellular and extracellular Mg2+ concentrations are almost equal? What mechanism is involved in managing Mg2+ after the initial influx, given the absence of Mg2+ pumps and major Mg2+ binding proteins? Follow-up studies demonstrated that MagT1-deficient cells have severely reduced basal intracellular Mg2+ concentrations (79). These results suggest that the effects seen on PLCy1 are dependent on general intracellular Mg2+ availability and further question the physiological role of variable fluxes that are proposed in the second messenger theory. Studying Mg2+ dynamics within the cell using fluorescent probes may help to draw definitive conclusions on this matter.
Cell Proliferation
Given its effect on RNA, DNA, and protein synthesis, Mg2+ is an important factor in the control of cell proliferation. Over the last 40 years, the role of Mg2+ in cell cycle control, protein synthesis, and growth factor response has been extensively studied, pioneered by several groundbreaking studies from the group of Harry Rubin (428, 430). Cell proliferation is largely dependent on protein synthesis, more than DNA or RNA synthesis. Inhibition of protein synthesis directly shuts down DNA synthesis, whereas there is a 2-h delay to achieve the same effect using RNA synthesis inhibitors (279). Protein synthesis is highly dependent on intracellular Mg2+ concentrations; increasing the Mg2+ content amplifies protein synthesis within 60 min, whereas DNA synthesis is only enhanced after 10 h (429, 514). Activation of proliferation is initiated by growth factors that increase glucose uptake and protein synthesis within minutes (225). Interestingly, Mg2+ is tightly regulated during these intracellular processes. Initial studies in cultured cells showed that applying insulin induced 20% higher intracellular Mg2+ concentrations after 16 h (445). Later studies with EGF using the fluorescent probe mag-fura 2 showed an impressive fourfold increase of intracellular Mg2+ from 0.3 to 1.4 mM after 20 min of epidermal growth factor (EGF) stimulation (199). The authors state that a rise in Mg2+ precedes DNA synthesis, but coincides with and thus may contribute to increases in protein synthesis. Recent studies identifying the molecular mediators of Mg2+-dependent cell proliferation have resulted in the membrane, magnesium, mitosis (MMM) model (432). The MMM model proposes that, upon growth factor binding, Mg2+ enters the cell or is released from phospholipids in the cell membranes (FIGURE 1). Increased cytosolic Mg2+ levels contribute to ribosomal activity and protein synthesis, eventually leading to DNA replication and mitosis. The mammalian target of rapamycin (mTOR) complex is a critical component of the MMM model, as it is the master regulator of cell cycle progression and proliferation (551). Growth factors binding to their receptors leads to phosphoinositide 3-kinase (PI3K) phosphorylation, which activates the mTOR complex (431, 548). Activation of mTOR is MgATP2~ dependent, and ATP has been suggested as the main regulator of mTOR activity. However, ATP levels do not change upon growth factor stimulation, whereas Mg2 + levels do (445, 537). Therefore, the MMM model proposes Mg2+ as the primary regulator of mTOR dynamics and cell proliferation.
Cancer
Tumor cells contain high concentrations of intracellular Mg2+ (508). In a mammary tumor cell line, Mg2+ can be transported into the cell even when extracellular Mg2+ concentrations were below physiological levels (203, 264, 566). Mg2+ uptake via divalent cation channel TRPM7 has been suggested to stimulate tumor cell proliferation (203, 264). TRPM7 expression is upregulated in hepatoma, pancreatic adenocarcinoma, gastric cancer, and breast cancer tissue (203, 278, 346, 581). Although TRPM7 has been primarily described as a Mg2+ channel, it is also permeable for other divalent cations (349). Given the involvement of Mg2+ in cell proliferation, the influx of Mg2+ via TRPM7 has been proposed as the main regulator of tumor growth. However, recent studies using prostate cancer cells suggest that TRPM7-mediated Ca2+ uptake may also play an important role in tumor growth (501). The expression of Mg2+ transporter CNNM3 is increased in human breast cancer tissue (217). CNNM3 binds oncogene PRL2 and facilitates the entry of Mg2+ in the tumor cell to drive cell proliferation. Elevated intracellular Mg2+ concentrations have been suggested to be beneficial for tumor growth because Mg2+ regulates several cancer-associated enzymes including telomerase and protein phosphatase 1D, which are involved in the glycolytic cycle and BER (74). However, the regulatory role of Mg2+ on these enzymes during the pathogenic state of tumor cell proliferation has never been investigated and, therefore, the exact role of Mg2+ in enzymatic regulation in cancer remains speculative.
In contrast to the proliferative phase of tumor growth, in which tumor cells have high intracellular Mg2+ concentrations, low intracellular Mg2+ concentrations are associated with increased rates of carcinogenesis and metastasis (74). Low Mg2+ conditions and impaired activity of DNA repair mechanisms reduces DNA protection against oxidative stress. Indeed, low dietary Mg2+ intake has been associated with the risk of several types of cancers. Epidemiological studies have established a correlation between low Mg2+ intake and colon cancer risk (160, 302, 534). In addition, in a study with 1,200 lung cancer patients and a similar number of controls, low dietary Mg2+ intake was associated with reduced lung cancer risk (326). However, these results could not be reproduced in other patient cohorts (325, 502).
REGULATION OF MAGNESIUM HOMEOSTASIS
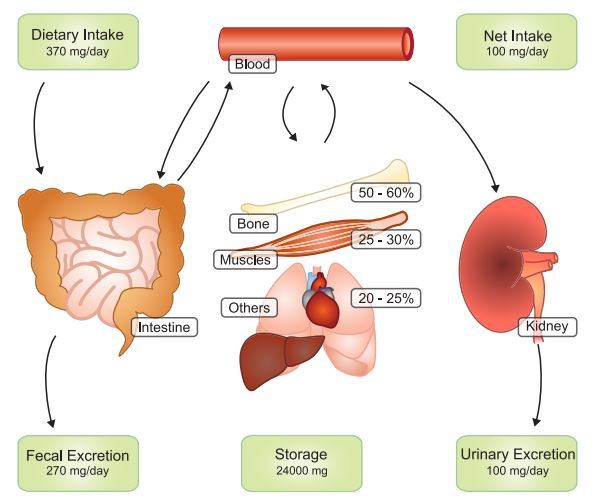
Mg2+ serum concentrations range between 0.7 and 1.1 mM in healthy people (323). To maintain constant plasma Mg2+ levels, the United States Food and Nutrition Board recommends a daily Mg2+ intake of 420 mg for men and 320 mg for women (1). Mg2+ homeostasis depends on the collaborative actions of the intestine, responsible for Mg2 + uptake from food, the bone, which stores Mg2+ in its hydroxy-apatite form, and the kidneys, regulating urinary Mg2+ excretion (FIGURE 2).
Figure 2
Magnesium in Intestine
Given a daily Mg2+ intake of 370 mg, ~30-50% is absorbed in the intestine, resulting in a net uptake of ±100 mg. However, if Mg2+ intake is low, early reports suggest that up to 80% of dietary Mg2+ can be absorbed (189). Mg2+ absorption in the gut depends on two separate pathways; paracellular transport is responsible for bulk Mg2+ absorption and takes place mostly in the small intestine, whereas fine-tuning occurs in the cecum and colon via transcellular transport (FIGURE 3). In spite of this, the intestine seems to have a limited role in regulation of the Mg2 + balance. In contrast to other minerals, intestinal Mg2+ absorption is poorly regulated and depends mainly on Mg2 + intake (216, 461). Thus the kidneys presumably primarily regulate the maintainance of Mg2+ homeostasis.
Small intestine
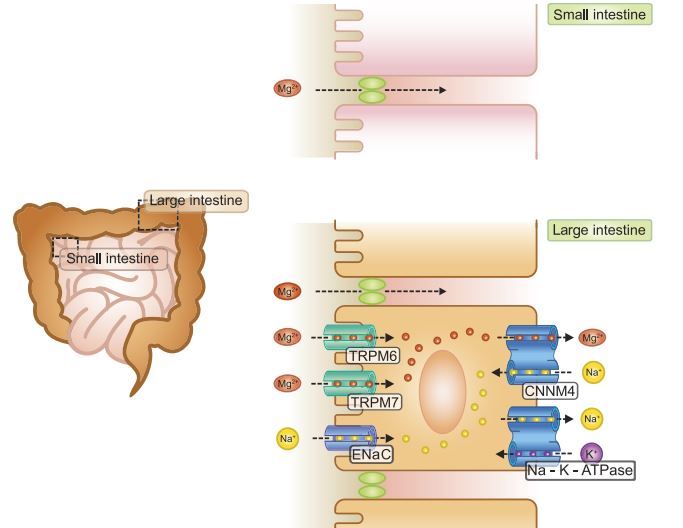
FIGURE 3 . Magnesium absorption in the intestine. Bulk Mg2+ is absorbed paracellular by the late part of the small intestine. Fine-tuning of Mg2+ absorption takes place transcellular by the colon, where TRPM6 and TRPM7 Mg2+ channels facilitate luminal Mg2+ uptake in the enterocyte. CNNM4 provides the basolateral Mg2+ extrusion mechanism. TRPM6, transient receptor potential melastatin type 6; TRPM7, transient receptor potential melastatin type 7; ENaC, epithelial sodium channel; CNNM4, cyclin M4.
Mg2+ absorption in the small intestine is hypothesized to be exclusively of a paracellular nature, since Mg2+ absorption in this region of the intestine correlates linearly to luminal Mg2+ concentrations (273, 409). Moreover, the epithelial Mg2+ channel TRPM6 is not expressed in the small intestine (196). Mg2+ is poorly absorbed in the duodenum, where unfavorable electrochemical gradients may even result in a limited amount of paracellular Mg2+ excretion (389). In more distal parts of the small intestine, such as late jejunum and ileum, the driving force for passive Mg2+ transport is established by the high luminal Mg2+ concentration and the lumen-positive transepithelial voltage of ~15 mV (164). The Km for Mg2+ transport in the distal small intestine has been reported to be in the range of 4-12 mM (343, 461). These results suggest that NaCl and water absorption are prerequisites for Mg2+ uptake, since water absorption concentrates luminal Mg2+. Tight junction permeability underlying paracellular Mg2+ transport is still
poorly understood. The small intestine is described as the most ion-permeable part of the intestine because of the relatively low expression of “tightening” claudins 1, 3, 4, 5, and 8 (20, 301). Claudins 16 and 19, which are linked to Mg2+ transport, are not expressed in the intestine (20,246). The exact composition of the tight junction complex facilitating intestinal Mg2+ absorption remains to be elucidated.
Large intestine
Mg2+ absorption in cecum and colon is thought to be transcellular of nature and is mediated by TRPM6 and TRPM7 on the luminal side of the enterocyte (FIGURE 3). Intestinal expression of TRPM6 is located in cecum and colon (196, 300). In a study with rat colon epithelium, 37% of Mg2 + was transported transcellularly (272). This suggests significant paracellular transport of Mg2+ in the colon, which would be unlikely given the expression of tightening claudins 3, 4, and 8 in this segment (301). In contrast to Ca2+, Mg2+ transport in colon is independent of 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] signaling, nor is TRPM6 expression dependent on 1,25(OH)2D3 (196, 272). It has been suggested that the basolateral Mg2+ extrusion mechanism of the enterocyte is coupled to the Na+ gradient (424). Indeed, the results of a recent study using CNNM4 KO mice suggest that CNNM4 may act as a Na+/Mg2+ exchanger at the basolateral membrane of enterocytes (575). CNNM4 KO mice suffer from hypomagnesemia, and functional analysis using Magnesium Green showed that CNNM4 overexpression increased Mg2+ efflux in HEK293 cells. However, patients with CNNM4 mutations do not suffer from hypomagnesemia (387, 402).
Magnesium in Bone
Approximately 50-60% of the total body Mg2+ content is stored in bone . Serum Mg2+ concentrations are closely related to bone metabolism; bone surface Mg2+ is continuously exchanged with blood Mg2+ (14). In bone, Mg2+ ions bind at the surface of the hydroxyapatite crystals. Mg2+ increases the solubility of Pi and Ca2+ hydroxyapatite and thereby acts on the crystal size and formation (443). Mg2+ induces osteoblast proliferation; therefore, Mg2+ deficiency results in decreased bone formation (320) (FIGURE 4) Mg2+deficient rats have reduced osteoblast numbers and decreased bone mass (434). Additionally, Mg2+ deficiency increases the secretion of proinflammatory cytokines such as tumor necrosis factor (TNF)-a, interleukin (IL)-10, and substance P (434, 552), all of which have been implicated in increased osteoclastic bone resorption (280). These effects may be further enhanced by reduced parathyroid hormone (PTH) and 1,25(OH)2D3 levels, which are often associated with hypomagnesemia (435). Interestingly, Mg2+-deficient rats have reduced chondrocyte column formation, which is associated with reduced SRY (sex determining region Y)box 9 (SOX9) expression (200) (FIGURE 4). SOX9 is a key transcription factor in chondrogenesis. In a recent gene expression study in the Mg2+-transporting segment of the kidney, SOX9 mRNA was the most increased in the lowMg2 + diet group (104). These findings suggest that SOX9 is an important transcription factor for bone and kidney Mg2+ homeostasis.
Magnesium in Kidney
Approximately 2,400 mg of Mg2+ is filtered by the glomeruli on a daily basis . The nephron recovers 95-99% of this; the remaining 100 mg leaves the body via the urine (FIGURE 2).
Proximal tubule
The mechanisms of proximal tubule (PT) Mg2+ reabsorption are poorly understood, but early micropuncture studies showed that ~ 10-25% ofMg2+ is reabsorbed by the proximal convoluted tubule segment of the nephron (304, 411). In the glomeruli, 70% of the serum Mg2+ is freely filterable, suggesting that the concentration in the glomerular filtrates and thus at the start of the PT ranges between 0.5-0.7 mM. The transepithelial potential difference ranges from slightly lumen negative ( 6 mV) in the early parts of the PT to positive (3 mV) in later parts (287). Micropuncture studies have shown that a 1.9 ratio between the concentrations of Mg2+ in the tubular fluid and the interstitial fluid is necessary to initiate Mg2+ transport (304). This finding could be explained by the poor tight junction permeability for Mg2+ in PT. As a result, water uptake via aquaporin 1 (AQP1) precedes Mg2+ reabsorption (410) (FIGURE 5). Consequently, Mg2+ reabsorption mainly occurs in the late parts of the PT, where the transepithelial chemical Mg2+ gradient is sufficient to favor Mg2+ transport. PT Mg2+ reabsorption is generally considered to be a passive paracellular process, but there might be some transcellular Mg2+ transport via a poorly characterized amiloride-sensitive mechanism (254). In both cases, sufficient Na+ transport is required to drive water transport that is a prerequisite for Mg2+ reabsorption. Hormonal effects on Na+ reabsorption in the PT will therefore also affect Mg2+ reabsorption in this segment. However, disturbances of proximal tubular Mg2+ reabsorption generally do not result in clinical symptoms, since more distal segments will compensate for reduced Mg2+ uptake in PT.
FIGURE 5 . Magnesium reabsorption in the kidney. The glomerulus filters the blood, and along the nephron 95% is reabsorbed. In the late proximal tubule (PT), Na+ and H2O reabsorption via NHE3 and AQP1 are prerequisites for paracellular Mg2+ transport. Approximately 10-25% of Mg2+ is reabsorbed in the proximal tubule. BulkMg2+ reabsorption (50-70%) takes place in the thick ascending limb of Henle's Loop (TAL). InTAL, Mg2+ reabsorption take place paracellular and depends on the uptake of Na+ and K+ via NKCC2. Fine-tuning (10%) of Mg2+ transport takes place transcellular in the distal convoluted tubule (DCT). In DCT, TRPM6 facilitates Mg2+ uptake from the pro-urine, which depends on the voltage gradient set by backleak of K+ via ROMK and K/1.1 potassium channels. At the basolateral membrane, Mg2+ is extruded via an unknown mechanism, which may be regulated by CNNM2 acting as Mg2+ sensor. Mg2+ extrusion depends on the Na+ gradient, set by the Na+-K+-ATPase. The activity of the Na+-K+-ATPase is in turn dependent on K+ recycling via Kir4.1. FXYD2 transcription encoding the y-subunit of the Na+-K+-ATPase is regulated by HNF1 0 and PCBD1. Regulation of Mg2+ transport in DCT depends on EGF and insulin. Upon activation of the EGFR and IR, an intracellular signaling cascade including PI3K, Akt, and Rac1 results in an increased TRPM6 membrane expression and increased channel activity. Additionally, estrogens have been shown to increase TRPM6 expression. PT, proximal tubule; TAL, thick ascending limb of Henle's loop; DCT, distal convoluted tubule; CNT, connecting tubule; CD, collecting duct; NHE3, Na+-H+ exchanger type 3; AQP1, aquaporin 1; NKCC2, Na+-K+-2CP cotransporter; ROMK, renal outer medulla K+ channel; ClC-Kb, chloride channel Kb; Kv1.1, voltage-gated K+ channel 1.1; TRPM6, transient receptor potential melastatin type 6; NCC, Na+-Cl~ cotransporter; CNNM2, cyclin M2; FXYD2, FXYD-domain containing 2; HNF1|8, hepatocyte nuclear factor 10; PCBD1, pterin-4 alpha-carbinolamine dehydratase 1; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; IR, insulin receptor; PI3K, phosphoinositide 3-kinase; Rac1, Ras-related C3 botulinum toxin substrate 1; Cdk5, cyclin-dependent kinase 5.
Thick ascending limb of Henle’s loop
Whereas most electrolytes are majorly transported in the PT, the thick ascending limb of Henle’s loop (TAL) is the main location for Mg2+ reabsorption (236, 299, 420). Due to the unique properaties of this segment, ~50-70% of filtered Mg2+ is reabsorbed here. Most of the Mg2+ is reabsorbed by the cortical part of the TAL, since medullary Mg2+ reabsorption is negligible (468). Paracellular bulk Mg2+ transport is dependent on the lumen-positive transepithelial voltage (+10 mV) that is determined by the activity of the Na+-K+-2Clcotransporter (NKCC2) and the subsequent secretion of K+ at the apical membrane (192). Inhibition of NKCC2 by furosemide diuretics therefore decreases TAL Mg2+ reabsorption (see sect. VB3). Further contributors to the transepithelial membrane voltage are K+ secretion via renal outer medullary potassium channel (ROMK) and paracellular backflux of Na+ ions as a result of decreasing luminal Na+ concentrations (329).
The reabsorption of Mg2+ in the TAL follows the paracellular pathway and therefore depends on the tight junction permeability (FIGURE 5). Tight junctions form a physical and chemical barrier between the epithelial cells. Their major components are proteins of the claudin family. Currently, 26 claudins have been described in humans (209). Tight junction permeability is determined by the individual claudins in each tight junction complex. TAL tubuli are known to express claudins 3, 10, 11, 14, 16, and 19. Claudins 16 and 19 are considered to be the main claudins influencing Mg2+ permeability, since mutations in these proteins result in renal Mg2+ wasting (293) (FIGURE 5).
However, the role of claudin 16 in Mg2+ reabsorption is controversial. Claudin 16 was initially considered to act as a paracellular Mg2+ channel, but this hypothesis has not been unequivocally confirmed (207,245). Reports using the claudin 16 knockdown (KD) mouse model and the LLCPK1 cell model suggest that claudin 16 increases Na+ permeability (227, 245, 466). This would imply that claudin 16 is mainly involv ed in the regulation of the transepithelial voltage gradient by controlling the paracellular Na+ backleak. In MDCK-C7 cells overexpressing claudin 16, Na + permeability yet remained stable, whereas Mg2+ permeability increased significantly (207). Claudin 16 KD mice demonstrate a twofold lower permeability ratio for Na + over Cl— without a change in paracellular conductance. Consequently, the transepithelial voltage collapsed, reducing the driving force for Mg2+ reabsorption in TAL (466).
Claudin 19 has been studied less extensively, but has been suggested to increase the tight junction barrier function (24). The claudin 19 KD mouse exhibits highly increased urinary excretion of K+, Mg2+, and Ca2+, but Mg2+ is the only electrolyte altered at the serum level (246). The discrepancy between studies with claudin 16 and claudin 19 isoforms might be explained by their interdependence in forming functional tight junction barriers (246, 247). Both in vitro and in vivo studies demonstrated that claudins 16 and 19 need to interact for proper insertion in the tight junction to become functionally active. Further differences in experimental results may depend on the endogenous expression of other claudin isoforms in the specific cell types used in these experiments.
Claudin 14 reduces the cation specificity of tight junction barriers, when coexpressed with claudin 16, or with claudin 16-claudin 19 complexes (180). Consequently, claudin 14 KO mice exhibit increased serum Mg2+ values and decreased urinary Mg2+ excretion. This agrees with previous findings in MDCK cells showing that claudin 14 acts as a nonspecific cation blocker (45, 560). Studies on claudin 14 KO mice have mainly focused on Ca2+ homeostasis, since claudin 14 expression is highly Ca2+ sensitive (121, 180). The CaSR regulates claudin 14 expression and Ca2+ reabsorption in the TAL by downregulation of two microRNAs, miR-9 and miR-374. Since CaSR is also activated by Mg2+, although to a lesser extent than Ca2+, it would be interesting to address the effect of elevated serum Mg2+ levels on claudin 14 expression in future studies.
Recently, claudin 10 has been identified as an important factor in cation selectivity in TAL, as demonstrated in a mouse model where claudin 10 was deleted specifically in this segment (57). Claudin 10 TAL-KO mice show hypermagnesemia, nephrocalcinosis, and impaired paracellular Na+ permeability. In the absence of claudin 10, TAL tight junctions became more permeable to Ca2+ and Mg2+ and the transepithelial voltage increased. These results are in line with in vitro studies overexpressing claudin 10b, suggesting that this splice variant is mainly expressed in TAL (57, 208).
Distal convoluted tubule
The distal convoluted tubule (DCT) determines the final urinary Mg2+ concentration, since no reabsorption of Mg2+ takes place beyond this segment. Approximately 10% of the total Mg2+ is reabsorbed by tightly regulated transcellular transport mechanisms (64). DCT cells form a high-resistance epithelium with a lumen-negative voltage of approximately —5 mV (192, 571). In DCT, TRPM6 divalent cation channels mediate luminal Mg2+ uptake (234, 235) (FIGURE 5). Within the kidney, TRPM6 is specifically expressed in DCT, and its activity is regulated by intracellular Mg2+ (540). TRPM6 contains six transmembrane spanning domains with a pore region between the fifth and sixth segment and a large kinase domain fused to the channel’s intracellular COOH terminus. TRPM6 may function in homo- and heteromeric tetramers with TRPM7, although there is some controversy about the necessity of TRPM7 for TRPM6 function (314, 588).
TRPM6 is regulated by numerous factors at the level of transcription, plasma membrane availability, and activity (69). EGF and insulin act on TRPM6 by a PI3K-Akt-Rac1 dependent mechanism, increasing the insertion of TRPM6 in the membrane (361, 515) (FIGURE 5). Insulin may directly affect TRPM6 activity through cylin-dependent kinase 5 (cdk5)-dependent phosphorylation of the channel. Patients with reduced EGFR or insulin receptor (IR) activity are therefore more susceptible to hypomagnesemia (361, 459). Additionally, estrogens increase TRPM6 mRNA expression (196). Over the last decade, several important interactors of TRPM6, including receptor for activated C-kinase 1 (RACK1) and prohibitin2 (PHB2/REA), have been identified (70, 71). RACK1 interacts with the a-kinase domain of TRPM6 in the autophosphorylated state, thereby reducing TRPM6 activity (70). Other modulators of TRPM6 activity include dietary Mg2+, pH, and ATP (516). Interestingly, acidification-induced current potentiation is dependent on residues p.Glu1024 and p.Glu1029, which also determine the pore selectivity for Mg2+ (313, 367). Moreover, recent findings indicate that TRPM6 is inhibited by low concentrations of intracellular ATP (IC50 29 M), questioning the physiological activity of monomeric TRPM6 channels (588). Extracellular ATP also inhibits TRPM6 activity via the purinergic receptor P2X4 (103).
A chemical gradient for Mg2+ entry in DCT cells is almost absent. The luminal Mg2+ concentrations vary between 0.2 and 0.7 mM, and the intracellular Mg2+ levels are typically in the range of 0.5-1 mM. Therefore, luminal Mg2+ entry is purely dependent on the negative membrane potential in the DCT cell. Luminal K+ channels are indispensable for maintaining the necessary driving force for Mg2+ uptake. The voltage-gated K+ channel Kv1.1 has been suggested to provide efflux K+ currents resulting in hyperpolarization of the luminal membrane, although expression levels in the DCT are limited (104, 176). To prevent Mg2+ overload and hyperpolarization of the luminal membrane, intracellular Mg2+ blocks Kv1.1 (179). Interestingly, recent studies suggest that other potassium channels in the luminal membrane of DCT cells may have comparable roles. ROMK is prominently expressed in DCT, and its expression in this segment is regulated by dietary Mg2+ (104, 572). Similar to Kv1.1, intracellular Mg2+ blocks ROMK currents, suggesting a regulatory function on Mg2+ homeostasis (578). Moreover, indirect inhibition of ROMK by aldosterone or eptihelial Na+ channel (ENaC) blockers represent the only effective approach to prevent renal Mg2+ wasting in most clinical situations (140).
Several proteins have been proposed to mediate Mg2+ extrusion to the bloodstream, but general consensus of the extrusion mechanism has not been reached (424). Due to the absence of a representative DCT cell model, the properties of Mg2+ extrusion have not been elucidated. Nevertheless, over the last decade several groups claimed to have identified Mg2+ extrusion proteins. Originally described in 2002, cyclin M2 (CNNM2, previously known as ACDP2) is exclusively expressed at the basolateral membrane of DCT and CNT cells within the kidney (105, 497, 545). Moreover, expression of CNNM2 is sensitive to dietary Mg2+ availability (104, 497). CNNM2 was initially depicted as Mg2+ transporter, since overexpression in Xenopus laevis oocytes allows uptake of a variety of divalent cations, with highest affinity for Mg2+ (184). However, these results could not be confirmed in mammalian cell lines (497). Alternatively, a Mg2+-sensing function has been proposed, since CNNM2 harbors a Mg-ATP binding site in its cystathionine- -synthase (CBS) domains (105). CNNM2 increases Mg2+ uptake in HEK293 cells (28). Nevertheless, it remains unclear whether CNNM2 mediates Mg2+ uptake directly or activates other Mg2+ carriers.
Recently mutations in the SLC41A1 Mg2+ transporter were described to cause a nephronophthisis-like phenotype (251). Immunohistological studies showed expression in DCT, but the stainings were not conclusive about the subcellular localization (apical or basolateral) of SLC41A1 proteins (251). By the use of Mag-Fura, SLC41A1 was demonstrated to increase both Mg2+ absorption and Mg2+ extrusion (251, 289). These results suggest that SLC41A1 plays a role in DCT Mg2+ reabsorption, although further studies are necessary to elucidate the mechanisms by which SLC41A1 mediates Mg2+ transport.
Parvalbumin is exclusively expressed in DCT within the kidney, where it may function as a Ca2+/Mg2+ buffer (456). Although parvalbumin has much higher affinity for Ca2+ than for Mg2+ (dissociation constants are ~5-10 nM for Ca2+ and ~30 M for Mg2+), the cation binding sites of parvalbumin will be mainly occupied by Mg2+ (377). This can be explained by the fact that the intracellular concentration of Mg2+ (0.5-1 mM) vastly exceeds that of Ca2 + (50-100 nM). In mouse and human kidney, parvalbumin is exclusively expressed in the early DCT (44). In late DCT and CNT, calbindin-D28K is the main Ca2+-binding protein. The exact role of parvalbumin in DCT remains to be investigated. Parvalbumin KO mice do not have altered serum or urine Mg2+ levels under basal conditions; these mice have reduced NCC expression, but display normal tubule morphology (44). DCT parvalbumin expression is highly sensitive to dietary Mg2+ availability, suggesting an important role for parvalbumin in Mg2+ reabsorption in DCT (104).
MAGNESIUM IN PHYSIOLOGY AND PATHOPHYSIOLOGY
The human body contains ~24g Mg2+, of which 99% is stored in bone, muscle, and other soft tissues. Mg2+ is critical to the function of basically every organ in the human body. Moreover, Mg2+ deficiency is associated with a wide range of diseases, and as a result Mg2+ supplementation is considered as potential treatment in many of them (TABLE 2). This part of the review focuses on the organ-specific functions of Mg2+ and provides an overview of all major diseases in which Mg2+ may play a role.
Magnesium in Brain
Low serum Mg2+ values are associated with a wide range of neurological diseases such as migraine, depression, and epilepsy. Neuronal Mg2+ concentrations are of major importance in the regulation of N-methyl-D-aspartate (NMDA) receptor excitability. NMDA receptors are essential for excitatory synaptic transmission, neuronal plasticity, and excitotoxicity and therefore play an important role in developmental plasticity, learning, and memory (384). NMDA receptors are activated upon glutamate binding and mediate the influx of Ca2+ and Na+ ions and the efflux of K+ ions. Every NMDA receptor consists of four subunits, each with different biochemical properties (97). In Mg2+ deficiency, NMDA receptors become hyperexcitable, which can be explained by inhibitory function of extracellular Mg2+ on the receptors (335, 372) (FIGURE 6). Glutamate from the presynaptic neuron will bind both the ionotropic 2-amino-3- (3-hydroxy-5-methyl-isoxazol-4-yl) propanoic acid (AMPA) and NMDA receptors on the postsynaptic neuron. At a normal membrane potential of —70 mV, Mg2+ ions block NMDA receptors. Therefore, only AMPA receptors will be activated and consequently facilitate an influx of cations. Only when the membrane potential rises above —60 mV the Mg2+ block is relieved and NMDA receptors are opened upon glutamate binding. The unlocking mechanism consists of a slow and a fast component, which depend on the relative expression of subunits that compose the channel (87, 536). Upon reduced extracellular Mg2+ concentrations, less NMDA channels will be blocked, and more NMDA channels can be opened at relatively low membrane potentials (351). This increased excitatory postsynaptic potential causes hyperexcitability of the neurons.
Table 2
In the adult brain, this process is further amplified by the action of inhibitory y-aminobutyric acid (GABA) receptors, whose function is also regulated by Mg2 + . GABAa receptors are ionotropic anion channels that open upon GABA binding and facilitate Cl— influx (250). Since the equilibrium potential of Cl— is 10-20 mV lower than the membrane potential, this influx contributes to hyperpolarization of the neuronal cells. Extracellular Mg2+ stimulates GABAA receptors resulting in hyperpolarized neuronal cells (353). When Mg2+ concentrations in the central nervous system (CNS) are low, GABAa receptors are less stimulated. Consequently, the membrane potential will be higher, which in turn relieves the Mg2+ block of the NMDA receptor and contributes to hyperexcitability of the neurons.
The final mechanism contributing to the hyperexcitability of NMDA-receptor rich neurons is inhibiting glutamate release from the presynaptic neuron. Release of glutamate can be inhibited by high extracellular Mg2+ concentrations (257, 318, 486). Although the exact mechanism by which Mg2+ reduces glutamate release is still unknown, it could be related to the inhibition of voltage-gated Ca2+ channels, as glutamate release is triggered by an influx of Ca2+ after an action potential (364).
As a result of the described mechanisms, low extracellular Mg2+ levels in the CNS contribute to the hyperexcitability of NDMA receptor. The excessive intracellular Ca2+ in the neurons may lead to the production of toxic reactive oxygen species (ROS) and eventually to neuronal cell death.
In addition to increasing the hyperexcitability of excitatory neuronal pathways, Mg2+ has an important role in the regulation of oxidative stress and the release of neuropeptides such as calcitonin gene-related peptide (CGRP) and substance P. CGRP is secreted from sensory neurons and has a vasodilatory effect (48). Mg2+ may increase CGRP expression and secretion, as has been shown in women with preeclampsia (26, 151), although an opposite effect was reported in women with Raynaud’s phenomenon (359). Mg2+ deficiency increases the release of substance P, which is a neuorinflammatory tachykinin (554), stimulating the secretion of inflammatory mediators such as IL-1, -2, -4, -5, -10, -12, and -13 as well as TNF-a (552, 554). Moreover, Mg2+ enhances the activity of nitric oxide synthases (NOS) through a NMDA receptor-dependent mechanism (80, 392). Nitric oxide (NO) has multiple functions in the brain including vasodilation, regulation of gene transcription, channel activity, and neurotransmitter release (493). Altogether, the role of Mg2+ in the regulation of neuropeptide release may have serious consequences in neuronal disease.
Migraine
Migraine has been linked to low levels of magnesium in serum and cerebrospinal fluid (CSF) (414, 457). Migraine headaches are the consequence of cortical spreading depression (CSD), which consists of an intense membrane depolarization and repolarization in neurons and glial cells (308, 388). CSD can be evoked by NMDA receptor activation (181). Therefore, patients with an increase in neuronal excitability due to low CSF Mg2+ levels are more susceptible to migraine attacks. Moreover, patients suffering from migraine are often hypotensive during attacks (462), which can be explained by increased NO levels. NO is an important vasodilator and modulator of brain blood flow (376). As an inhibitor of NO production, reduced Mg2+ values may result in decreased NO levels (80, 392).
The first reports of Mg2+ treatment for migraine patients appeared in the 1960s and 1970s (542), and since then the pharmacological role of Mg2+ has been slowly recognized. Although presently the effectiveness of Mg2+ treatment for the majority of patients is still debated (334, 386), Mg2+ is a second line drug for migraine patients (148, 237). Over the last decades several double-blind placebo-controlled randomized trials have provided evidence for a beneficial effect of oral Mg2+ supplementation on the number of migraine attacks (294, 393, 507) as well as the intensity of the pain during these attacks (149, 294). One clinical trial failed to show any favorable effect (397), but patients in this study suffered from diarrhea. Although Mg2+ is generally well tolerated, supplementation with certain Mg2+ salts often results in diarrhea and malabsorption of the Mg2 + . Intravenous Mg2+ supplementation in acute migraine and cluster headache treatment provided pain relief in several cases (47,112, 332, 333). In contrast, other studies failed to demonstrate such an effect (78). A recent meta-analysis combining five studies on the effect of intravenous Mg2+ administration on migraine did not show significant improvements on pain relief [Risk ratio (RR): —0.07, 95% confidence interval (CI): —0.23 to 0.09]. However, the relatively low number of patients included (n = 295) is one of the major limits of this analysis (86). Efficacy of Mg2+ treatment in combination with other drugs is doubtful; Mg2+ combined with metoclopramide and riboflavin did not demonstrate any efficacy in treating migraine (94, 328).
Depression
In 1921, Weston (557) reported the beneficial role of Mg2 + in treatment of patients with depression. Nevertheless, large-scale, placebo-controlled double blind clinical trials assessing the efficacy of Mg2+ supplementation on depression are still lacking. Studies examining the association between serum Mg2+ concentrations and depression severity are not conclusive; some studies report altered blood Mg2+ levels, while others do not find differences (reviewed in Ref. 114). Since serum Mg2+ levels do not necessarily reflect neuronal Mg2+ availability, determining Mg2+ levels in CSF may be more relevant for patients with depression. Only three cross-sectional studies have addressed this issue, and none of them found altered CSF Mg2+ concentrations in depressed patients compared with healthy controls (35, 171, 310). Despite this, several investigations have proposed that Mg2+ may relieve depression by blocking the NMDA receptor, whose dysfunction is a major causative factor in depression pathology (135, 143, 400, 557). To date, two interventional studies have investigated the role of Mg2+ in treating depression and their results are contradictory. In a randomized trial examining depressed elderly patients with diabetes mellitus type 2 and hypomagnesemia, Mg2+ supplementation was as effective as standard imipramine treatment (37). However, this study lacked a placebo control group. Moreover, it should be noted that imipramine therapy is nowadays largely replaced by selective serotonin reuptake inhibitors (SSRI). Another limit of the studies is the small sample population (23 patients). Therefore, large-scale studies are necessary to delineate a role of Mg2+ in the treatment and prevention of depression.
Epilepsy
Seizures are often associated with genetic and acquired forms of hypomagnesemia (see sect. V). Many studies have found that patients suffering from epilepsy display lower blood Mg2+ values (212, 375, 482). The link between Mg2+ status and the development of seizures may be explained by the role of Mg2+ in NMDA receptor blockade. Most studies addressing this issue show a small but significant decrease in CSF Mg2+ levels in epilepsy patients (492). In eclampsia patients, Mg2+ has proven to be successful in reducing the risk of recurrent convulsions (198). For other types of seizures, the evidence is less conclusive. In 1933 the first report of Mg2+ infusions in eight status epilepticus patients, a life-threatening form of epilepsy in which patients suffer from continuous seizures without regaining consciousness, was successful in all cases (496). However, modern reports of Mg2+ infusion treatment of status epilepticus patients have more variable outcomes and are not as conclusive (159, 383). Mg2+ infusions are therefore considered a second line of treatment, when anti-epileptic drugs and anesthetics have proven to be unsuccessful. Absence of large-scale randomized double blind placebo-controlled trials obstructs the implementation of Mg2+ as general antiepileptic treatment (9).
Stroke
Stroke is one of the major causes of death in the Western society and has been associated with a drop in serum Mg2+ levels (19). There may be multiple roles for Mg2+ in the etiology of stroke. Low serum Mg2+ levels increase NMDA receptor activity and thus more glutamate and Ca2+ influx. Excessive Ca2+ and glutamate influx via the NMDA receptor may be the basis of excitotoxicity during stroke (137). Since clinical trials with NMDA receptor antagonists have proven to be unsuccessful in treatment of stroke (256), it is, however, unlikely that NMDA receptor blockade alone can fully explain the role of Mg2+ in the development and onset of stroke. Mg2+ also blocks other voltage-gated Ca2+ channels that may be involved in Ca2+ cytotoxicity. Additionally, Mg2+ has a vasodilatory effect, which may be beneficial for patients suffering from ischemic stroke. Although more than 100 neuroprotective agents were tested in animals, not a single agent has been proven successful in a phase 3 clinical trial (111). After several pilot studies showed beneficial effects of Mg2+ on clinical outcome parameters, two large randomized controlled trials have been performed to determine the role of Mg2+ administration in stroke treatment (355, 356, 449). The Intravenous Magnesium Efficacy in Stroke Trial (IMAGES) enrolled over 2,500 stroke patients and gave a 16 mmol MgSO4 bolus injection within 12 h of a stroke, followed by a maintenance dose of 65 mmol over 24 h. No beneficial effects were reported on the primary outcome, death and disability at 3 mo [odds ratio (OR) 0.95, 95% CI 0.80-1.13]. However, in a subgroup of patients treated within the first 3 h (3.3% of the cohort), a favorable death or disability outcome of 0.66 (95% CI 0.25-1.70) was reported (356). Since animal studies indicate that Mg2+ treatment is only successful when applied within 3 h of the onset of the stroke (579), a largescale follow-up randomized controlled trial is currently running which aims to treat patients with Mg2+ within the first hours after stroke in a prehospital and emergency department setting. Although the trial is still running, first reports indicate that they have succeeded in including 72% of ~ 1,000 patients within the first hour (448). Final results from this FAST-MAG study will help determine the efficiency of Mg2+ treatment for stroke patients.
Approximately 5% of all strokes are caused by subarachnoid hemorrhage (SAH) that results from ruptured aneurysms. Delayed cerebral ischemia (DCI) is the major cause of death and disability in patients that survive the first 24 h (126). Interestingly, patients with SAH often present with hypomagnesemia (531, 532). Vasoconstriction is the main cause of DCI, and this may be enhanced when the patient is Mg2+ deficient. Over the last decade, several clinical trials have examined the addition of Mg2+ administration to the standard nimodipine treatment (358, 533, 555, 568). In 2005, in the Magnesium and Acetylsalicylic acid in Subarachnoid Hemorrhage (MASH-I) study poor outcomes were 23% reduced (RR 0.77; 95% CI 0.54-1.09) and DCI was reduced by 35% (RR 0.65; 95% CI 0.40-1.05) (533). However, more recent studies in larger study-cohorts did not report beneficial effects. In the intravenous magnesium sulfate for aneurysmal subarachnoid hemorrhage (IMASH) double-blind randomized placebo-controlled trial, 6 mo favorable outcome was similar between the patients given MgSO4 and the control, 64 and 63%, respectively (RR 1.0; 95% CI 0.7-1.6) (568). Comparable results were obtained more recently, in the MASH-II multicenter randomized placebo-controlled in a cohort of 1,200 patients; 74% had a favorable outcome in the Mg2+ group and 75% in the control group (RR 1.03; 95% CI 0.85-1.25) (124). Overall, these studies suggest that intravenous Mg2+ treatment does not improve clinical outcome after aneurysmal subarachnoid hemorrhage.
Brain injury
Mg2+ deficiency is regularly found in patients with traumatic brain injury (TBI) and spinal cord injury (SCI) (81, 268). Reduced CSF Mg2+ levels increase oxidative stress (ROS, NO) and lipid peroxidation, which both contribute to the severity of TBI (77, 416). Additionally, it has been proposed that Mg2+ deficiency increases the release of substance P in TBI, resulting in neuronal cell death and edema (539). In animal experiments, Mg2+ improved sensorimotor/motor function as well as cognitive function (233). In a small study of 30 TBI patients, Mg2+ supplementation improved patient outcomes as measured by the Glasgow outcome scale (OR 4.13; 95% CI 1.39-12.27) (116). A large phase 3 randomized placebo-controlled clinical trial tested the effect of two doses of Mg2+ treatment on 6 mo mortality, seizures, functional measures, and neuropsychological tests in 500 TBI patients. Surprisingly, patients receiving Mg2+ treatment did significantly perform worse on the primary outcome than control patients (48 vs. 54; 95% CI — 10.5 to —2), suggesting an adverse effect of Mg2+ (511). A meta-analysis on all clinical trials with TBI patients confirmed that there is no evidence for a neuroprotective role of Mg2+ in TBI (25). This demonstrates again the difficulties in translating the results obtained in animal studies to the clinic (111).
Parkinson’s disease
Parkinson’s disease is characterized by a loss of dopaminergic neurons. Parkinson’s patients have low Mg2+ concentrations in cortex, white matter, basal ganglia, and brain stem (580). Interestingly, rats with chronic low Mg2+ intake exhibit a significant loss of dopaminergic neurons (381). In vitro experiments often use differentiated PC12 cells and 1-methyl-4-phenylpyridium ion (MPP + ) to model Parkinson’s disease at the cellular level. In this experimental model, mitochondrial Mg2+ concentrations were decreased as was demonstrated using the mitochondrial KMG-301 fluorescent Mg2+ probe (475). Moreover, Mg2+ transporter SLC41A1 is located on the PARK16 locus that is associated with Parkinson’s disease (576). Recent characterization of the SLC41A1-pA350V single nucleotide polymorphism (SNP) linked to Parkinson’s disease evidenced a gain-of-function effect (290). These studies suggest that Mg2+ supplementation may be beneficial for patients suffering from Parkinson’s disease.
Other brain pathologies
Low serum Mg2+ levels have also been associated with a wide range of neurological pathologies including schizophrenia, bipolar disorder, neuroses, addiction, stress, and Alzheimer’s disease (538). Although this suggests that Mg2+ deficiency plays a role in the etiology of these pathologies, all reports to date are of an epidemiological nature. There are currently no reports of clinical trials examining the effect of Mg2+ supplementation on the disease outcome of these diseases.
Magnesium in Lung
Dietary Mg2+ intake has been repeatedly associated with lung function, as assessed by forced expiratory volume (FEV) and forced vital capacity (FVC) (60, 174). Lung Mg2+ research suffers from a lack of fundamental studies. As a result, the mechanisms that explain the role of Mg2+ in lung function are poorly understood, and hypotheses are mainly based on studies in other cell types and organs. Nevertheless, the role of Mg2+ in lung function may be explained at three levels: 1) Mg2+ has a strong vasodilator and bronchodilator effect; 2) Mg2+ regulates the release of acetylcholine (ACh) and histamine; and 3) Mg2+ acts as anti-inflammatory agent (FIGURE 7).
From studies on coronary artery related diseases, it is known that Mg2 + has a vasodilatory effect (512,513). Like many vasodilators, Mg2+ also has a bronchodilating effect (229, 374). Although the mechanisms underlying Mg2+- induced bronchodilation remain to be elucidated, Mg2+ is known to inhibit the release of ACh and histamine, both known to induce bronchoconstriction (88, 291, 398, 427). Moreover, Mg2+ may reduce the airway inflammation that underlies several lung diseases, including chronic obstructive pulmonary disorder (COPD) and cystic fibrosis. In line with this, Mg2+ deficiency has been reported in children with bronchitis, and low Mg2+ levels can induce an inflammatory response in lung allografts (43, 440). Most of our understanding of the role of Mg2+ in inflammation comes from studies in brain, heart, and intestine (450, 509, 553), and only a few studies have examined the anti-inflammatory function of Mg2+ in lung. Nevertheless, it is generally accepted that Mg2+ protects against inflammation by reducing oxidative stress, inhibiting substance P release, and preventing Ca2+ toxicity by inhibiting voltage-gated Ca2+ channels (450,553). Mg2+ also modulates NFkB activation by influencing lipid peroxidation (17, 153). All together, these characteristics make Mg2+ a potential therapeutic agent for lung diseases such as asthma and COPD.
Asthma
Several studies have reported low serum Mg2+ levels or low erythrocyte Mg2+ levels in asthmatic patients (21, 141, 219). However, others could not detect a Mg2+ deficiency in patients with asthma, suggesting that Mg2+ levels may depend on the severity of the disease (109, 276). Since Mg2+ relaxes smooth muscle cells, low Mg2+ levels cause bronchoconstriction and vasoconstriction, resulting in more asthmatic exacerbations (374). Moreover, Mg2+ regulates the release of ACh and histamine, which have both been implicated in asthma (170, 371). Interestingly, asthmatic C57/Bl6 mice have lower serum and intracellular Mg2+ concentrations than controls, which could be explained by a decreased renal TRPM6 expression (265). In 1940, Victor Haury (221) was the first to treat bronchial asthma patients with Mg2+ injections to relieve asthmatic paroxysms. Since then, ~25 randomized controlled studies have been published examining the effects of nebulized and intravenous Mg2+ administration in asthma patients. A recent systematic review failed to demonstrate significant improvement of respiratory function [standardized mean difference (SMD) 0.17, 95% CI —0.02 to 0.36] or hospital admissions (RR 0.68, 95% CI 0.46 to 1.02) for patients with acute asthma who had been given nebulized Mg2+, although both parameters almost reached statistical significance (348). In studies using intravenous Mg2+ injection, respiratory function increased slightly in adults, but the most significant improvements were found in children (SMD 1.94, 95% CI 0.80-3.08), and children’s hospital admissions were reduced (RR 0.70, 95% CI 0.54-0.90) (348). A recent systematic review within the Cochrane collaboration addressing nebulized Mg2+ for treatment of acute asthma concluded that respiratory function is not significantly improved in Mg2+-treated patients compared with patients receiving 2-agonists (404). However, the patients covered by this systematic review were mainly adult patients. Recently, the outcomes of the MAGNEsium Trial In Children (MAGNETIC) randomized controlled study were published, showing improvement of the asthma severity score at 60 min (0.25, 95% CI 0.02-0.48) and 240 min (0.20,95% CI 0.01-0.40) after inhalation of MgSO4 (405). The MAGNETIC study was not published at the time of the systematic reviews, and future analysis including this trial may further substantiate the beneficial effects of Mg2+ in treatment of children with asthma.
Cystic fibrosis
Mg2+ deficiency has been repeatedly reported in patients with cystic fibrosis (210, 380). Patients with cystic fibrosis are often treated with recombinant human DNase-I to degrade the viscous mucus. However, the recombinant DNase requires Mg2+ to function, and efficiency of the treatment depends, therefore, on the Mg2+ status of the patient (444). It was suggested that Mg2+ treatment in itself would be sufficient to trigger endogenous DNase activity in the sputum (426). To further examine the potential role of Mg2+ as treatment for cystic fibrosis patients, a small double-blind, randomized, placebo-controlled crossover study tested the effect of oral Mg2+ supplementation on respiratory muscle strength and disease severity (444). Patients supplemented with Mg2+ increased their maximal inspiratory pressure (MIP) and maximal expiratory pressure (MEP). Although these first results are promising, more large-scale follow-up studies are necessary to assess the effects of Mg2+ in cystic fibrosis patients.
COPD
Serum Mg2+ levels have been associated with disease progression in patients with COPD (31, 46). Given the bronchodilating effects of Mg2+, several studies have examined whether intravenous or nebulized Mg2+ may benefit COPD patients. In 1995, a small study of 27 COPD patients examined the effect of 1.2 g MgSO4 infusion after -agonist administration. Although the sample size was small, Mg2+- treated patients demonstrated higher peak expiratory flow values compared with placebo-treated patients (22 ± 29 vs. 6 ± 24%) (485). In another small randomized double-blind controlled trial, Mg2+ infusion resulted in improved functional respiratory capacity (—0.48 l, 95% CI: —0.96, —0.01), inspiratory capacity (0.21 l, 95% CI: 0.04-0.37), MIP (10 cmH2O, 95% CI: 1.6-18.4), and MEP (10.7 cmH2O, 95% CI: 0.20-21.2) (123). In contrast, a recent study using combined intravenous and nebulized MgSO4 administration in 62 COPD patients did not detect significant effects on the primary outcome, as measured by hospital admission, intubation, and hospital death rates, nor did they find improved lung function (370). A larger recent study testing nebulized MgSO4 did not detect improved lung function after 90 min, as determined by FEV (136). Patients that received 151 mg MgSO4 in addition to standard salbutamol treatment demonstrated similar FEV ( — 0.0261,95% CI —0.15 to 0.095). Altogether, the efficacy of Mg2+ treatment in COPD remains unclear and may depend on the route of administration and the combination with the use of additional drugs.
Magnesium in Heart and Vasculature
Mg2+ plays an important role in heart function by influencing myocardial metabolism, Ca2+ homeostasis, vascular tone, peripheral vascular resistance, and cardiac output. Mg2+ exerts its effects in three ways: 1) Mg2+ regulates the activity of ion channels in the cardiac cells, thereby affecting the electrical properties of the myocardium (354); 2) Mg2+ regulates myocardial contractility by influencing the intracellular Ca2+ mobility; and 3) Mg2+ has an anti-inflammatory and vasodilatory effect (FIGURE 8).
The cardiac action potential consists of five phases: phase 0 is the rapid depolarization by the influx of Na+. Phase 1 consists of rapid repolarization by efflux of K+. Phase 2, named the plateau phase, is the longest phase and marks Ca2+ entry. Phase 3 allows final repolarization of the cell by restoration of the membrane potential. Phase 4 is the stable phase with a resting potential of ±90 mV (190). Mg2+ is mainly important in phases 2 and 3 of the myocardial action potential, exerting its effect on K+ and Ca2+ channels. In phase 2, Mg2+ inhibits L-type Ca2+ channels (Cav1.2) to prevent Ca2+ overload and cell toxicity (558). Mg2+ can bind a COOH-terminal EF hand motif of the channel and thereby influences the Ca2+ current (63). The effects of Mg2+ on the current through the L-type Ca2+ channels (ICaL) may depend on the channel’s phosphorylation state, since phosphatase treatment decreases the inhibitory effects of Mg2+ (547). In phase 3, delayed rectifier K+ channels repolarize the cell by rapid-activating (IKr) and slow-activating (IKs) currents. High [Mg2+i] inhibits IK currents in frog and guinea pig cardiomyocytes (128, 563). This effect probably depends on the slow-activating component of the current, since rapid-activating currents seem insensitive to Mg2+ inhibition (499). The intracellular block of inward rectifier K+ channels Kir2.1 and Kir2.2 by Mg2+ substantially influences phase 3 and phase 4 of the action potential (556). This block of the IK1 current is relieved by high extracellular K+ concentrations (56).
In recent years an increasing amount of attention has been directed to the role of [Mg2+i] in cardiac excitation-contraction coupling (342). Mg2+ has often been considered as a natural Ca2+ antagonist, since it can compete with Ca2+ for binding sites in proteins and Ca2+ transporters (257). The effect of Mg2+ on cardiomyocytes is mainly explained by its role of Ca2+ mobilization. Mg2+ binds calmodulin, troponin C, and parvalbumin, and therefore a reduced [Mg2+i] may result in alterations in the unbound Ca2+ fraction (15). Mg2 + may also affect the main Ca2+-transporting proteins in the cardiomyocytes. Mg2+ acts as substrate in a complex with ATP for cardiac Ca2+-ATPases and alters the affinity of Na+-Ca2+ exchanger type 1 for Ca2+ (NCX1) (55, 58, 342). There is a dearth of physiological studies on the effect of Mg2+ on NCX1 and SERCA activity, and available studies mainly rely on modeling and in vitro experiments. Nevertheless, tight regulation of [Mg2+i] in cardiac cells is necessary for optimal cardiac function. This is substantiated by the fact that high [Mg2+i] can cause cardiac arrest, and by the impressive capacity of cardiomyocytes to maintain constant [Mg2+i] (506, 521).
An important role of Mg2+ in heart and vasculature function is a substantial vasodilatory effect that has been reported in animal and human studies (18, 161, 512, 513). Although the results from animal studies suggest that Mg2+-induced vasodilation is due to the regulation of NO synthesis, human studies show that it is independent of NO activity (512, 513). Moreover, Mg2+ deficiency promotes oxidative stress notably in endothelial cells (118, 561), resulting in increased reactive oxygen species (ROS) and cytotoxicity (590). In contrast, high Mg2+ in the cell increases eNOS activity and suppresses the synthesis of vasoconstrictor endothelin-1 (277, 327). In conditions of low Mg2+- induced oxidative stress, the endothelium develops a state of permanent inflammation, which is marked by increased NFkB activitiy (153). NFkB is the master regulator of transcription of cytokines and pro-inflammatorty genes, including IL-1a. As a result of this local inflammation, the vessel wall will recruit monocytes and trigger the proliferation and migration of vascular smooth muscle cells. These processes are facilitated by the increased expression of matrix metalloproteases 2 and 9 in low Mg2+ conditions (153, 382). Eventually low Mg2+ concentrations may, therefore, result in atherosclerosis, vascular calcifications, or thrombosis.
Coronary artery disease
Over the last 20 years, an increasing number of studies have demonstrated that low serum Mg2+ levels and low Mg2+ intake are associated with an increased risk of coronary artery disease (CAD), artherosclerosis, and metabolic syndrome (8, 222, 316, 587). Low serum Mg2+ levels have been associated with a higher mortality risk in CAD patients (162). There may be several ways in which Mg2 + supplementation benefits patients with CAD. Since Mg2 + has a strong anti-inflammatory role, Mg2+ results in an improved lipid profile, reduced free oxygen radicals, and improved endothelial function (317, 490). Mg2+ prevents blood clotting by reducing platelet aggregation (437), and it has a strong vasodilator effect (512, 513). These properties make Mg2+ an important factor in the development and management of CAD. Mg2+ improves several aspects of vascular function in CAD. Reduced serum Mg2+ concentrations are associated with an increase in carotid intimamedia thickness and risk for sudden cardiac death (218, 324, 391). In a randomized, double-blind, placebo-controlled study of 50 CAD patients, oral Mg2+ supplementation ameliorated endothelial function (472). Moreover, Mg2+ reduced platelet-induced thrombosis in a randomized prospective, double-blind, crossover, and placebo-controlled study in 42 CAD patients (471). Six months of Mg2+ supplementation increased maximal oxygen uptake (Vo2 max) and left ventricular ejection fraction (LVEF) in 53 CAD patients (401). Taken together, these studies suggest that Mg2+ levels should be closely monitored in CAD patients and propose Mg2+ as potential drug to improve quality of life in CAD patients.
Myocardial infarction
In the 1970s, a pioneering paper by Abraham et al. (9) associated myocardial infarction with a significant drop in serum Mg2+. Indeed, low serum Mg2+ levels have been associated with an increased risk of acute myocardial infarction (AMI) (484). The role of Mg2+ in preventing myocardial infarction may be caused by relaxing endothelial and smooth muscle cells in the heart and vasculature (18, 161, 512). Moreover, heart rate variability is a risk factor for AMI and Mg2+ may prevent arrhythmia (474, 484). Several studies have addressed the effect of Mg2+ on myocardial infarction. In the 1980s, studies reported a 20% reduction of infarct size in Mg2+-treated patients, and decreased mortality after Mg2+ infusion (352, 415). Several follow-up studies suggested that decreased rates of arrhythmias after infarction explain the lower mortality (76, 487). After these initial promising results, several clinical trials have addressed this subject. The Leicester Intravenous Magnesium Intervention Trial 2 (LIMIT-2) included 2,316 AMI patients and found 24% reduced mortality after 28 days in the Mg2+ group (95% CI: 1-43%) (569). Moreover, two smaller studies by Shechter and colleagues reported reduced mortality rates of 50 and 40% (469, 470). Contrary to these findings, in the fourth Infarct Survival and Magnesium in Coronies (ISIS-4) study, a randomized factorial trial in 58,050 patients showed no beneficial effects of intravenous Mg2+ administration on survival (4). The results from the ISIS-4 study were called into question because the statistical analysis did not reflect the heterogeneous studies used in the analysis, the low mortality in the placebo-group was indicative of a low-risk comparison group, and the late time-point of Mg2+ administration, namely, after and not during reperfusion, was at odds with animal data demonstrating the effectiveness of Mg2+ (463). However, also a second large-scale randomized double-blind study with 6,213 patients and an earlier time point of Mg2+ infusion (3.8 h compared with 8 h in ISIS-4) also failed to show a decrease on 30-day mortality rates (2). Indeed, a recent meta-analysis within the Cochrane collaboration concludes that there is no beneficial effect of Mg2+ on mortality in AMI patients (OR 0.99, 95% CI 0.94 to 1.04) (312). However, it should be noted that the ISIS-4 study provides 72% of the power in this analysis and that the Mg2+ doses used within the analyzed studies differ significantly. Nevertheless, current guidelines do not recommend Mg2+ administration in AMI patients.
Arrhythmia
In 1935, Dr. Zwillinger was the first to report an antiarrhythmic effect of Mg2+, and since then sporadic reports of patients treated with Mg2+ have appeared in the literature (595). However, the field suffers from a lack of largescale randomized controlled trials, and therefore the exact clinical benefit of Mg2+ in treatment of arrhythmia remains to be determined. Mg2+ is known to have a function in regulating cardiac K+ and Ca2+ channels, so it affects the cardiac action potential. As a result, hypomagnesemia in itself has been proposed as a cause of arrhythmia, specifically in combination with stress or alcoholism (362, 524). Clinical studies have demonstrated that treatment success strongly depends on the type of arrhythmia (232). Atrial fibrillation is one of the most common and dangerous complications after cardiac surgery. In the last few decades, several small-scale studies have examined the effect of Mg2+ in preventing these fibrillations (93). Meta-analysis of these studies concluded that Mg2+ infusion may prevent atrial fibrillations (201, 474). Therefore, the European Association for Cardiothoracic Surgery and the Canadian Cardiovascular Society recommend prophylaxis with intravenous MgSO4. However, recent reports point to problems associated with many studies at the level of double-blinding, primary outcome, and intention-to-treat analysis. When only high quality studies are included, Mg2+ does not have a preventive effect on atrial fibrillations (OR: 0.94, 95% CI: 0.61-1.44) (93). A similar discrepancy between low- and high-quality studies has been noted for studies addressing the effects of Mg2+ on supraventricular arrhythmias (108). Interestingly, for treatment of torsades des pointes, Mg2+ has been implemented as a first line of treatment after several studies in the 1980s showed beneficial effects (395, 527, 528). However, due to the absence of large-scale clinical trials, optimal doses for treatment are still under debate (211, 243, 244). Other arrhythmias, including refractory ventricular fibrillation and monomor- phic ventricular tachycardia, are insensitive to Mg2+ treatment (150, 220).
Preeclampsia
Since the 1950s, intravenous MgSO4 administration has gradually become the standard treatment for preeclampsia and eclampsia, and nowadays the treatment is widely advocated by the World Health Organization (223,407, 594). The mechanisms of action underlying the effect of Mg2+ in the treatment of these patients are largely unknown. Mg2 + may reduce preeclampsia by its effect as vasodilator in the vasculature, but it cannot be excluded that Mg2+ also functions as an anticonvulsant through blockade of the NMDA receptor and reduces cerebral edema (147). Recent Cochrane systematic reviews showed that MgSO4 treatment in preeclamptic women reduced the risk for eclampsia by >50% (RR 0.41, 95% CI 0.29-0.58), and there was a trend towards lower maternal mortality (RR 0.54, 95% CI 0.26-1.10) (130). MgSO4 demonstrated similar ratios for reduced risk for eclamptic convulsions comparable to anticonvulsant medication, with 59% compared with diazepam (RR 0.41, 95% CI 0.29-0.58) and 66% compared with phenytoin (RR 0.34, 95% CI 0.24-0.49) (131, 132). Moreover, a recent meta-analysis of “real-world” use of MgSO4 for the treatment of preeclampsia confirmed the results from the clinical trials and showed ~50% reduction of eclampsia risk in most studies (337). Interestingly, although Mg2+ was successful in preventing eclampsia, it did not change the risk of death or disability for children at 18 mo (RR 1.06, 95% CI 0.90-1.25) or the risk of death or disability for women at 2 yr (RR 0.84, 95% CI 0.60-1.18) (5,6).
Hypertension
Low serum Mg2+ levels are frequently linked to high blood pressure (30, 267, 270). Intracellular Mg2+ may reduce the intracellular Ca2+ concentration within vascular smooth muscle cells. This Mg2+-induced vasodilation is thought to be the mechanism by which Mg2+ alters the blood pressure. Moreover, high extracellular Mg2+ reduces the endothe- lin-1 expression and causes an increase in prostacyclin (PGI2) levels, contributing to vasodilation (27, 59, 303, 447). Additionally, Mg2+ inhibits the production of NO (392). Mg2+ was first used to lower blood pressure in 1925, when Mg2+ infusion was found to lower blood pressure by reducing the peripheral vascular resistance (53). Several studies have shown that oral Mg2+ intake reduces both systolic and diastolic blood pressure (260, 275, 559); however, other studies fail to see such an effect (72, 152). A systematic review of the Cochrane Hypertension Group reported a small reduction of diastolic blood pressure (DBP; -2.2 mmHg, 95% CI -3.4 to -0.9), but not of the systolic blood pressure (SBP; -1.3 mmHg, 95% CI -4.0 to 1.5) (119). Another meta-analysis detected a small but dose- dependent effect of Mg2+ on blood pressure, for each 10 mmol/day —4.3 mmHg SBP (95% CI —6.3 to —2.2) and -2.3 mmHg DBP (95% CI —4.9 to 0.0) (262). However, the outcomes may be biased by the inclusion of poor quality studies that tend to overestimate the effects. In contrast, a meta-analysis of a subset of studies, including patients on antihypertensive drugs with high blood pressure (SBP >155 mmHg) reports much stronger effects of oral Mg2+ treatment on SBP ( — 18.7 mmHg, 95% CI —14.95 to —22.45) and DBP ( — 10.9 mmHg, 95% CI —8.73 to —13.1) (425). These results suggest that Mg2+ may be beneficial for certain subgroups of hypertensive patients and that comparing the highly heterogeneous studies may underestimate the effects of Mg2+ in these groups.
Vascular calcification
Vascular calcification is frequently observed in patients suffering from chronic kidney disease (CKD) (452). Calcifications are a major contributor to cardiovascular death that accounts for 50% of all deaths in CKD (522). Vascular calcification is the consequence of a disturbed mineral metabolism including increased serum Pi levels. Increased serum levels of FGF23 and PTH enhance the formation of calicification. Low serum Mg2+ levels are associated with vascular calcification, and hemodialysis patients with higher serum Mg2+ levels show higher survival (258, 259). Although the mechanisms of action are not completely understood, there are two contributing factors: 1) Mg2+ prevents the formation and deposition of Ca/P nanocrystals and the development of appatite structures (83); and 2) Mg2+ inhibits the transdifferentiation of the smooth muscle cells in the vessel well into osteoblast-like cells (282). In both processes Mg2+ prevents vascular calcifiction, and thus hypomagnesemic patients are at risk. Therefore, Mg2+ supplementation has been proposed as Pi-binder to reduce vascular calcification in CKD patients. Several studies have shown that combined administration of Ca2+ and Mg2+ is as effective as standard treatment options (252). In the recent CALMAG study comparing MgCO3/Ca(OAc)2 with Sevalamer-HCl in 200 hemodialysis patients, both treatments were effective in reducing serum Pi levels without increasing Ca2+ levels (106).
Magnesium in Muscle
Mg2+ mainly exerts its effects on skeletal muscle function as a Ca2+ antagonist on Ca2+-permeable channels and Ca2+- binding proteins. Muscle contraction is a highly Ca2+-dependent process, initiated Ca2+ release from the sarcoplasmic reticulum. Ca2+ binds to troponin C and myosin to induce the conformational changes of these proteins that will result in contraction (193). Mg2+ competes for the Ca2+-binding sites on these proteins. Although the affinity of troponin and myosin for Mg2+ is much lower than for Ca2+ (292), the effects of Mg2+ are not negligible (239, 403). In the resting state, Mg2+ is present in concentrations 10,000 times higher than Ca2+ in muscle cells (292); therefore, Mg2+ will occupy all Ca2+ binding sites, and it is only after Ca2+ is released from the sarcoplasmic reticulum that Mg2+ is replaced. However, in Mg2+-deficient conditions, not as much Ca2+ is required to displace Mg2+, resulting in hypercontractibility, which presents as muscle cramps and spasms in the clinic.
Moreover, as a cofactor of ATP, Mg2+ availability is essential for the function of the ryanodine receptor (RyR), which allows rapid Ca2+ release from the ER, and Ca2+-ATPase of sarcoplasmic reticulum (SERCA), which mediates the return of Ca2+ to the SR after contraction. Intracellular Mg2+ and Ca2+ concentrations determine how many ATP molecules bind to the RyR, which determines the opening of these channel receptors and the release of Ca2+ (117). SERCA also depends on Mg2+ as cofactor of ATP. Additionally, Mg2+ can bind to the Ca2+ binding pocket of SERCA (584). The role of Mg2+ in the regulation of the Na+-Ca2+ exchanger (NCX) has been poorly studied so far.
Muscle cramps
Muscle cramps are a recurrent and prominent symptom in patients with severe/chronic hypomagnesemia (49, 215). Although the role of Mg2+ in the pathogenesis of muscle cramps is not completely understood, it is hypothesized that Mg2+ directly influences muscular contractions by antagonizing Ca2+-binding proteins. Moreover, Mg2+-deficient patients may suffer from neuronal hyperexcitability that can contribute to muscular contraction. Mg2+ has consequently been considered as treatment for muscle cramps in several studies. However, there is little evidence that Mg2 + may relieve muscle cramps in the general population. A recent Cochrane systematic review and meta-analysis of all published studies shows no significant reduction in the number of cramps after Mg2+ treatment ( — 3.93%, 95% CI: —21.12 to 13.26%) (168). In contrast, a Cochrane review from 2002 demonstrated that Mg2+ might be beneficial for muscle cramps in pregnancy (OR 0.18, 95% CI 0.05-0.60) (583). Both meta-analyses are limited by a relatively small patient population. Large-scale studies are necessary to ascertain the utility of Mg2+ for specific subpopulations or disease-related muscle cramps.
Magnesium in Pancreas
Mg2+ has been implicated in both endocrine and exocrine functioning of the pancreas. In the pancreatic acini, intracellular Mg2+ antagonizes Ca2+-activated signaling events and enzyme secretion (573). It is known that ACh and cholecystokinin 8 (CCK8) evoke an increase in intracellular Ca2+, which in turn activates calmodulin, resulting in the phosphorylation of proteins on the enzyme-containing vesicles. These vesicles will migrate towards the plasma membrane for exocytosis and secretion. Interestingly, ACh and CCK8 activation cause a marked Mg2+ efflux, allowing Ca2+ signaling to occur (483, 564). Its antagonizing role may be explained by inhibition of Ca2+ transporting proteins such as SERCA, PMCA, and Ca+-ATPases (120,584).
In the islets of Langerhans, Mg2+ may influence the secretion of insulin, although experimental results are conflicting as to whether insulin secretion is increased or decreased. Milner and Hales (344) reported that Mg2+ reduced insulin secretion in an ex vivo model using rabbit pancreas. These results were confirmed in rat pancreas and rat insulinoma cells (99, 357). However, in rats, one study found increased insulin secretion in Mg2+-deficient rats, while other studies in Mg2+-deficient rats did not report alterations in plasma insulin concentrations (202, 338,418). In addition, patients with low serum Mg2+ levels show decreased insulin secretion (422). The discrepancies among different experiments may be explained by the Ca2+ availability in each of the models. Since the effect of Mg2+ on insulin can be explained by its Ca2+ antagonizing role, it may not depend on the Mg2+ concentration itself, but on the cytosolic Ca2+/Mg2+ ratio.
Diabetes
Patients with diabetes mellitus type 2 often have low serum Mg2+ levels (36, 491, 541). These low serum levels are associated with poor disease outcome and may even increase mortality (98). Hypomagnesemia may contribute to the development of diabetes mellitus type 2 by increasing insulin resistance. Insulin receptors (IR) are part of the family of tyrosine kinase receptors, and the kinase function is dependent on the binding of two Mg2+ ions (249). Upon activation of the IR, a complex intracellular signaling cascade is activated and mediated via insulin receptor substrate proteins (505). In low Mg2+ conditions, activation of the IR may result in diminished signal transduction, contributing to insulin resistance. Studies with hypomagnesemic rats bear this out, as lower IR phosphorylation was detected, although differences between individual organs were reported (419, 498). It has also been proposed that increased expression of other effectors such as IL-1, IL-6, IL-8, TNF-a, norepinephrine, epinephrine, and ROS may contribute to insulin resistance in Mg2+ deficiency (206). Interestingly, common SNPs in the TRPM6 gene are associated with an increased risk of developing diabetes mellitus type 2 (489). TRPM6 cannot be activated by insulin when these SNPs are present (361). These results suggest that Mg2+ levels may influence the onset and development of diabetes mellitus type 2. Several studies have examined the clinical effects of oral Mg2+ supplementation on glycemic control in diabetes mellitus type 2 patients. Some of these studies demonstrate impressive effects in reducing glucated hemoglobin (HbA1c) levels and fasting glucose concentrations (385, 423), but other studies show no improvement of glycemic control (107, 204). A meta-analysis of 8 studies, including a total of 370 patients, evidenced a reduction of fasting glucose levels ( — 0.56 mM; 95% CI, —1.10 to —0.01), reflected in a nonsignificant (P = 0.1) reduction of HbA1c levels (—0.31%, 95% CI, —0.81 to 0.19) (488). All together, these results indicate that Mg2+ supplementation may be a promising avenue for achieving glycemic control in diabetes patients.
Magnesium in Liver
The role of Mg2+ in the liver is poorly studied. However, given that many of the enzymatic reactions that take place in the hepatocytes are dependent on Mg2+, particularly in fat metabolism, the importance of Mg2+ should not be underestimated. Mg2+ supplementation has been reported to reduce alanine aminotranferase (ALT) levels in obese women with hypomagnesemia (421). However, this result could not be reproduced in normomagnesemic patients, although a lower dose of Mg2+ was used in this study (271). The first reports of hypomagnesemia in liver diseases such as cirrhosis and nonalcoholic fatty liver disease suggest that liver function contributes to proper intestinal Mg2+ absorption (286, 526). Currently, the first clinical trials are being initiated to test the effects of Mg2+ supplementation in patients with liver cirrhosis.
Magnesium in Immunity
Mg2+ is considered as an anti-inflammatory agent that reduces the expression and release of substance P and other proinflammatory molecules. Mg2+ also influences acquired immunity by regulating the proliferation and development of lymphocytes (156). Deletion of the TRPM7 Mg2+ channel caused cell death in the chicken B cell line DT40, which could be partially rescued by culturing the cells in high Mg2+ containing medium (455). In a mouse model with a specific T-cell deletion of TRPM7, T lymphocyte development was blocked at the CD4—CD8— stage, resulting in decreased CD4+ and CD4 + CD8 + cells in the thymus (266). Moreover, mutations in the MagTl Mg2+ channel are causative for immunodeficiency and have been associated with decreased CD4+ T lymphocyte levels (311). These results suggest that Mg2+ is essential for T lymphocyte development and proliferation.
1. X-linked T-cell immunodeficiency
Patients with X-linked immunodeficiency with Mg2+ defect, Epstein-Barr virus infection and neoplasia (XMEN) have mutations in MagTl (79, 311). They present with chronic Epstein-Barr virus infections, low CD4+ T-cell counts, and defective T-lymphocyte activation. These effects are hypothesized to result from a loss of PLCy1 activation due to reduced Mg2+ influx via MagT1 (311). Recent studies in asthma patients confirm the importance of Mg2+ availability for CD4+ function (315). An increased risk in T-cell lymphoblastic leukemia has been associated with Mg2+ deficiency (441). However, the role of Mg2+ in T-cell signaling needs to be investigated before further conclusions can be drawn. Special attention should be given to the involvement of other Mg2+ carriers, since TRPM7-deficient T cells seem protected from Fas-receptor-induced apoptosis (115).
Magnesium in Bone
Bone hydroxyapatite structures mainly consist of Pi and Ca2+ and are bound by Mg2+ ions at the surface of the hydroxyapatite crystals. Mg2+ increases the solubility of the minerals and thereby acts on the crystal size and formation (443). Crystals in Mg2+-deficient bone are larger, and the bone may therefore be brittle and more susceptible to fractures (89). Moreover, Mg2+ stimulates osteoblast proliferation, suggesting that Mg2+ deficiency results in decreased bone formation (320) (FIGURE 4). The role of bone in Mg2+ homeostasis is described in more detail in section HLB.
Osteoporosis
Several studies have associated low serum Mg2+ values with osteoporosis (61,213). Although most studies have been performed in postmenopausal women, there is some evidence of low bone Mg2+ content in elderly subjects. In this study the patients had normal serum Mg2+ levels, but displayed significantly increased retention in a loading/tolerance test (91). A few small-scale studies have examined the effects of oral Mg2+ supplementation (200-750 mg/day) on bone mineral density (BMD) in patients with osteoporosis. In a pioneering study in 1991, daily administration of 600 mg Mg resulted in an 11% increased BMD after 12 mo (10). However, many other supplements including 500 mg/day Ca were simultaneously used, making it difficult to distinguish the effects of Mg2+. Subsequently, multiple studies have examined the effect of Mg2+ supplementation in different populations (138, 436, 494). Mg2+ seems to increase BMD in all of the studies, although the effects are relatively small (1-3%) and the small study sizes limit the conclusions that can be made from them. Testing the effect of Mg2+ supplementation in large cohorts of osteoporosis patients may further establish Mg2+ supplements to treat osteoporosis.
DISTURBANCES OF MAGNESIUM HOMEOSTASIS
Over the last decade, clinical interest in Mg2+ has been growing. Mg2+ deficiency has been associated with a wide range of diseases including diabetes mellitus type 2, hypertension, migraine, and depression. Therefore, the Mg2+ balance in patients is clinically significant; however, Mg2+ status in patients is not routinely determined. When Mg2+ is assessed in patients, total serum Mg2+ is the most commonly measured parameter. However, total serum Mg2+ levels do not necessarily represent body Mg2+ availability, since ionized Mg2+ determines the bioactive fraction. Only 2% of the clinical laboratories in the United States offer ionized Mg2+ tests, thus leaving total serum Mg2+ as the most used clinical representative of the patient’s Mg2+ status (139).
However, serum Mg2+ levels represent <1% of the body Mg2+ content, and therefore, it is a poor predictor of the body Mg2+ status. Additionally, the Mg2+ concentration in red blood cells is higher than in the serum (1.65-2.65 mM), and extra care should be taken to prevent hemolysis, which can result in a misrepresentation of total serum Mg2+ (520). In a recent systematic review, red blood cell (RBC) Mg2+ content has been proposed as an alternative measure to determine Mg2+ status, since its value is strongly affected by alterations in dietary Mg2+ intake in six studies (n = 130) (565). Based on the limited changes of ionized serum concentration after dietary Mg2+ supplementation or depletion, the authors reject ionized Mg2+ as marker for patient Mg2+ status. However, the rationale that alterations in dietary Mg2+ intake necessarily result in a physiologically relevant effect in Mg2+ availability is questionable, since the kidney and bone have a large capacity to compensate for reduced Mg2+ absorption. Only after a long-term depletion, patients may develop a clinically relevant hypomagnesemia.
However, the opposite also holds true; patients may be severely Mg2+ deficient, although serum or RBC Mg2+ levels are normal. In this case, the Mg2+ concentrations in bones and soft tissues are severly decreased, after long-term compensatory Mg2+ release to keep serum Mg2+ levels within normal range. To identify such patients, a Mg2 + loading test has been proposed in which acute oral Mg2 + administration is used to assess Mg2+ retention (363). In this test, the serum Mg2+ concentration and fecal Mg2 + excretion are calculated to determine intestinal Mg2+ absorption. Mg2+-deficient patients will have lower bone Mg2+ content and therefore show high Mg2+ retention (90). Thus the loading test allows quantification of the exchangeable pool of Mg2+, which is more sensitive than serum Mg2+ concentrations. However, this test is rarely used in clinic, and as a result, standardization is lacking. Within a research setting, a urine excretion of <60% of the Mg2+ load is generally considered normal.
If a Mg2+ disturbance is suspected, urinary Mg2+ concentrations are regularly determined. It should be noted that reliable urine Mg2+ determinations require at least a complete 24-h sampling, since the circadian rhythm influences renal Mg2+ excretion (158). The results of urine Mg2+ tests may provide information about the cause of Mg2+ deficiencies; normal or high urinary excretion indicates renal Mg2+ wasting, whereas low Mg2+ excretion suggests reduced intestinal absorption. This information can then be used to guide treatment plans.
Importantly, physicians must be aware that urinary Mg2+ concentrations are poor indicators of a patient’s Mg2+ status when kidney function is reduced. In several pathophysiological circumstances such as diabetes and chronic kidney disease, filtration may be altered. Additionally, the use of certain drugs including diuretics may cause bias in determining a patient’s Mg2+ status.
Hypermagnesemia
Serum Mg2+ levels above 1.1 mM are generally considered hypermagnesemic. Hypermagnesemia may be clinically observed in patients suffering from nausea, vomiting, lethargy, headaches, and/or flushing. When Mg2+ levels rise above 3.0 mM it may even cause severe cardiac defects that are characterized by brachycardia, hypotension, and prolongation of the QRS, PR, and QT intervals (TABLE 3). Extreme hypermagnesemia can therefore result in coma, asystole, and death by cardiac arrest. However, hypermagnesemia is rare and until now no genetic causes for it have been identified. Hypermagnesemic patients are often treated by infusion of Ca2+ salts to antagonize the cardiac effects of Mg2+ (433). Also the efficacy of hemodialysis in the treatment of acute hypermagnesemia has been illustrated (228, 379).
Table 3. Symptoms of Mg2 + disturbances
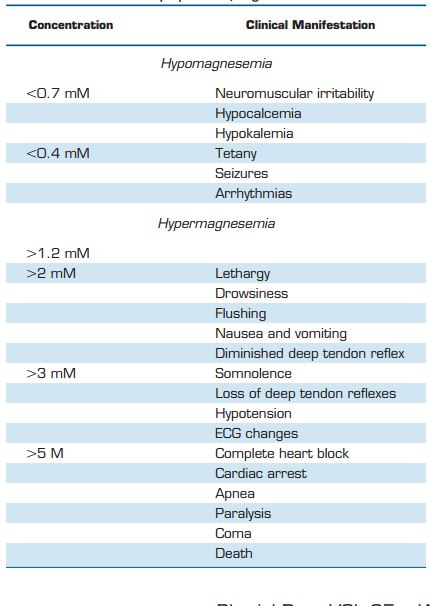
1. Drug-induced hypermagnesemia
Only a few drug-induced cases of hypermagnesemia have been reported, and most of them are the direct consequence of the administration of Mg2+ itself or Mg2+-containing drugs (TABLE 4).
epsom salts. Epsom salts consist of MgSO4 and are generally used as bath salts or a home remedy against abdominal pain, constipation, and muscle strains. Excessive ingestion of Epsom salts may result in Mg2+ overdose and hypermagnesemia. As a result, several fatal cases have been reported in literature (52, 231).
cathartics, laxatives, and enema . Several Mg2+ derivates have been used as cathartics, laxatives, and enemas with the similar goals of softening the stool and easing defecation by increasing their water content. For a long time magnesium citrate [Mg3(C6H5O7)2] was the most commonly used cathartic. Due to the risk of developing hypermagnesemia and other electrolyte disturbances, polyethylene glycol and electrolyte lavage solutions are currently the first drugs of choice (378, 451). Several Mg2+-containing substances [Mg3(C6H5O7)2, Mg(OH)2, MgSO4] have been used as laxatives. Mg2+ substances elevate the intestinal osmotic pressure, but also act on aquaporin-3 expression and thus increase water permeability (373). Mg2+ has been used as a component of enemas to treat constipation. However, both use of Mg2+ as a laxative and as an enema may result in fatal hypermagnesemia (413, 521, 586). Therefore, Mg2 + administration should be avoided in patients with reduced kidney function, and serum Mg2+ should be closely monitored during treatment.
Hypomagnesemia
Hypomagnesemia is generally defined as serum Mg2+ levels below 0.7 mM. Patients suffer from nonspecific symptoms such as depression, tiredness, muscle spasms, and muscle weakness, and diagnosis therefore may take years (TABLE 3) (177, 523). Only severe Mg2+ depletion (<0.4 mM) may lead to cardiac arrhythmias, tetany, and seizures. Secondary to hypomagnesemia, disturbances in K+ and Ca2+ handling are often detected. Hypokalemia can be attributed to increased renal K+ secretion via ROMK in the connecting tubule (CNT) and collecting duct (CD) (248). Low intracellular Mg2+ levels release the Mg2+-dependent inhibition of ROMK channels, resulting in increased renal K+ secretion. Hypocalcemia can be explained by low PTH levels due to altered activation of the CaSR (574).
Hypomagnesemia is generally treated by oral Mg2+ supplementation (±360 mg/day), although oral Mg2+ intake may cause diarrhea at high doses. Intravenous Mg2+ supplementation may be more effective, but this treatment has the disadvantage that it requires regular hospital visits. The treatment regimen of intravenous Mg2+ supplementation normally consists of 8-12 g of magnesium sulfate in the first 24 h followed by 4-6 g/day for 3 or 4 days (523). When serum Mg2+ levels are extremely low or are accompanied by hypokalemia, Mg2+ supplementation may not be sufficient to restore normal Mg2+ levels. In that case, patients are often cosupplemented with K+ or receive amiloride to prevent K+ secretion.
The following section of this review will focus on the causes of hypomagnesemia. Drug-induced and genetic hypomagnesemia will be distinhished from more general origins of Mg2+ deficiency (TABLES 4 AND 5).
General causes of hypomagnesemia
a) dietary mg2+ intake. Estimations state that up to 60% of American do not meet daily Mg2+ requirements (163, 281). Chronic inadequate intake of Mg2+ leads to hypomagnesemia. Consumption of Mg2+-rich foods such as kelp, nuts, green vegetables, and whole grains may prevent this. However, insufficient Mg2+ intake may also be caused by pathological conditions such as anorexia nervosa (51).
vomiting and diarrhea. Vomiting and diarrhea may further exacerbate the effects of inadequate Mg2+ uptake. Specifically, diarrhea is the consequence of inadequate water reabsorption along the intestine. Since water reabsorption is a prerequisite for Mg2+ reabsorption to set the concentration gradient, diarrhea may result in Mg2+ deficiency.
alcoholism . Since the early 1960s it has been recognized that alcoholism may cause severe hypomagnesemia (224, 336). Patients suffer from unexplained renal Mg2+ wasting, but may also have reduced intestinal Mg2+ absorption due to vomiting or diarrhea. in hepatocytes, ethanol completely blocked Mg2+ uptake (75). A similar mechanism may take place in the kidney, explaining reduced Mg2+ reabsorption. Alcoholics often have reduced PTH levels, which may further contribute to low serum Mg2+ levels (7).
Table 4. Drug-induced Mg2+ disturbances
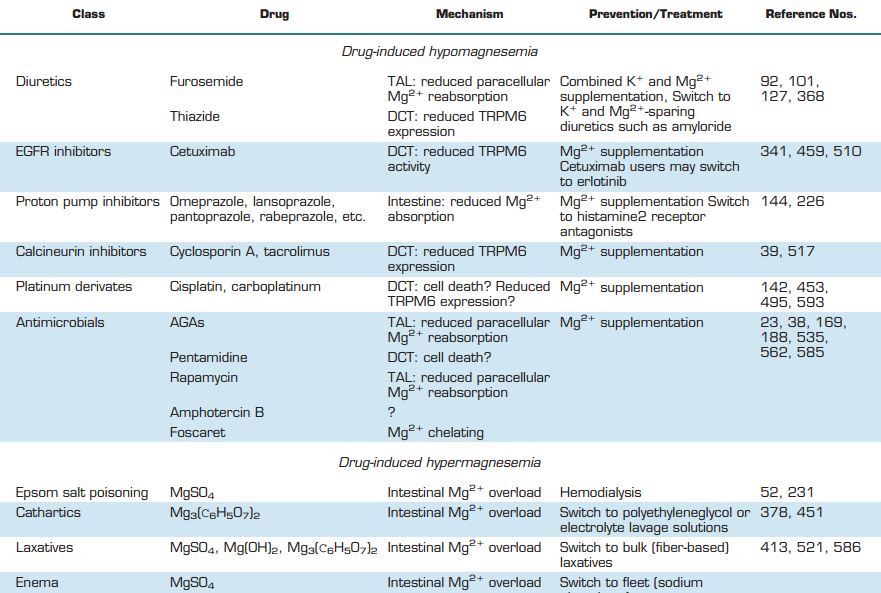
Table 5. Genetic causes of hypomagnesemia

Genetic hypomagnesemia
a) cldn16. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis type I (FHHNC type I; OMIM 248250) is caused by mutations in claudin 16, previously known as paracellin-1 (480). Patients suffer from renal Mg2+ wasting, hypomagnesemia, renal Ca2+ wasting, renal parenchymal calcification (nephrocalcinosis), and renal failure. Serum and urinary Na+, K+, Cl_, and HCO3 values are initially normal, but may be indirectly affected after progression of renal failure. Sometimes these symptoms are extended to urinary tract infections, kidney stones, and hyperuricemia; Mg2+ supplementation is not capable of restoring normal serum Mg2+ levels or slowing disease progression (567). A few dozen different mutations have been reported, all characterized by a recessive mode of inheritance (177). All symptoms can be traced to the TAL, the main site for paracellular Ca2+ and Mg2+ reabsorption. Claudin 16 is part of the tight junction between the cells, and disruption of these tight junctions results in a lack of Ca2+ and Mg2+ reabsorption in TAL, which can only partially be compensated in the downstream DCT and CNT segments.
cldn19. Similar to FHHNC type I, patients with FHHNC type 2 (OMIM 248190) suffer from severe hypomagnesemia accompanied by hypercalciuria and nephrocalcinosis. Additionally, patients have ocular defects consisting of macular colobomata, significant myopia, and horizontal nystagmus. FHHNC type 2 is caused by mutations in claudin 19, which is expressed in the TAL segment of the kidney, in parallel with claudin 16. In the initial publication from Konrad et al. (293), 12 patients from 10 families were genotyped and characterized. Remarkably, 9 of 12 patients developed chronic kidney disease or underwent kidney transplantation. Indeed, other studies confirmed that FHHNC type 2 patients are more prone to developing of CKD and develop the disease at an earlier age compared with type I patients (177). Over the years, several treatment regimens have been proposed, including oral magnesium supplementation, thiazide diuretics, and indomethacin. However, none of these treatments significantly increased serum Mg2+ values (177).
TRPM6. Hypomagnesemia with secondary hypocalcemia (HSH; OMIM 602014) is characterized by extremely low serum Mg2+ levels (0.1-0.3 mM) accompanied by low serum Ca2+ levels, which result in severe muscular and neurological complications including seizures and mental retardation (454, 543). The disorder was first characterized by Paunier and colleagues in 1968 and later mapped to a region at chromosome 9q in 1997 (390, 544). In 2002, two independent groups identified mutations in TRPM6 to be causative for HSH (454, 543). TRPM6 forms the epithelial Mg2+ channel responsible for transcellular Mg2+ transport in the colon and DCT segment of the kidney (540). Therefore, mutations result in reduced intestinal absorption and renal Mg2+ wasting. HSH has an autosomal-recessive mode of inheritance, and currently a few dozen mutations have been found. Patients are generally treated with Mg2+ supplements and antiepileptic drugs against seizures. Serum Mg2+ levels improve after supplementation, but did not recover to normal levels (297).
egf. Isolated autosomal recessive hypomagnesemia (IRH; OMIM 611718) is caused by mutations in the EGF gene (197). In a consanguine family from Dutch origin, two sisters presented with serum Mg2+ levels of 0.53 and 0.56 mM and urinary Mg2+ values of 3.9 and 3.7 mmol/24 h, respectively (173). Serum Ca2+, Na+, K+, CU, HCO7, and blood pH values were normal. The patients presented with epileptic seizures during their first year of life, which could be controlled by antiepileptic drugs. Moreover, psychomotor retardation was observed in these patients. Plasma renin activity, plasma aldosterone, and parathyroid hormone concentrations were in the normal range. Homozygosity mapping and subsequent Sanger sequencing of gene candidates led to the identification of a homozygous c.C3209T mutation in exon 22 resulting a p.P1070L missense mutation at protein level (197). This residue is particularly important for plasma membrane targeting of the EGF molecule and the mutation results in impaired basolateral sorting of pro-EGF. Therefore, TRPM6 activity is not stimulated, resulting in renal Mg2+ wasting (197, 515). Until now, only a single family has been described with EGF mutations, but studies with EGFR antagonists further underline the clinical importance of EGF for renal Mg2+ handling (discussed in detail in sect. VB3).
kcna1. In a Brazilian family, mutations in KCNA1 encoding voltage-gated K+ channel Kv1.1. cause autosomal dominant hypomagnesemia (OMIM 176260) (176). The patient presented in the clinic with muscle cramps, muscle, weakness, tetanic episodes, and tremor. Serum Mg2+ values were low (0.37 mM), whereas other electrolytes and metabolites including Na+, K+, Ca2+, Pi, uric acid, bicarbonate, urea, creatinine, glucose, bilirubin, aminotransferases, alkaline phosphate, and lactate dehydrogenase were all normal. Urinary creatinine clearance as well as Mg2+ and Ca2 + excretion were within normal range. KCNA1 mutations were previously linked to ataxia and myokymia (62, 146). Therefore, a cerebral MRI was performed in these patients, showing slight atrophy of the cerebral vermis. Family members suffer from myokymic discharge in electromyograph analysis, which is in line with the previously observed mixed phenotype. Intravenous Mg2+ infusion improved the clinical symptoms. Kv1.1 has been proposed to cause apical hyperpolarization, which allows the uptake of Mg2+ via TRPM6. The p.N255D (c.A763G) mutation identified in the Brazilian family disrupts Kv1.1 activity and therefore may reduce the driving force for Mg2+ transport. Although many KCNA1 mutations have been reported, even in residues very close to the p.N255, none of these has yet been associated with hypomagnesemia, even though Kv1.1 function is impaired (12, 263). Identification of additional hypomagnesemic families with Kv1.1 mutations may aid our understanding of Kv1.1 function in DCT. It has been suggested that other factors contribute to the apical membrane potential and may compensate for a loss of Kv1.1 function; ROMK may be one of the compensatory factors (104,140).
cnnm2. Mutations in CNNM2 are causative for hypomagnesemia with seizures and mental retardation (HSMR; OMIM 613882). Two unrelated families with seizures and dominant hypomagnesemia were reported to carry CNNM2 mutations (497). In these patients, serum Mg2 + levels range between 0.3 and 0.5 mM, but no other electrolyte disturbances were detected. The patients’ symptoms include seizures, loss of consciousness, loss of muscle tone, headaches, and staring (340). Recently, five additional families were reported (28), making CNNM2 the most common genetic cause of isolated hypomagnesemia after TRPM6 and CLDN16-19. Interestingly, in this new cohort CNNM2 mutations were linked to a phenotype of impaired brain development and mental retardation. This intellectual disability was most prominent in a family with a recessive pattern of inheritance, emphasizing the heterogeneous inheritance of CNNM2 depending on the location and severity of the mutations. HSMR patients are treated with antiepileptic drugs and Mg2+ supplements. Serum Mg2+ levels improved after supplementation, but did not reach normal levels. Although the exact function of CNNM2 remains to be elucidated, mutations can disrupt the MgATP binding domain and reduce CNNM2 membrane expression (28, 105). Recent data from patch-clamp and Mg2+ uptake experiments favor the hypothesis that CNNM2 does not transport Mg2+ itself, but rather regulates other Mg2+- transporting proteins (28, 105, 497).
kcnj10. Mutations in KCNJ10 encoding the Kir4.1 K+ channel can cause seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance/epilepsy, ataxia, sensorineural deafness, and renal tubulopathy syndrome (SeSAME/EAST; OMIM 612780) (54, 417, 458). Patients were reported to have marked electrolyte abnormalities, including hypokalemic metabolic alkalosis without hypertension, severe hypomagnesemia, and renal Na+, K+, and Mg2+ wasting. In some patients, high renin and aldosterone levels, salt craving, and polyuria were observed. Kir4.1 is hypothesized to be involved in K+ recycling at the basolateral membrane of DCT cells. When Kir4.1 is mutated, K+ availability becomes rate limiting for Na+-K+-ATPase activity. Thus the Na+-K+-ATPase will be inhibited, resulting in a reduced potential across the basolateral membrane. Consequently, Na+ and Mg2+ transport will be reduced in DCT. To compensate for this, ENaC activity in CNT will be increased at the expense of K+ excretion via ROMK. Therefore, SeSAME/EAST patients suffer from severe hypomagnesemia and hypokalemia. To treat the hypomagnesemia, patients are often given Mg2+ and K+ supplements, in combination with aldosterone antagonists or ENaC inhibitors (34). SeSAME/EAST patients suffer from a severe neurological phenotype consisting of tonic-clonic seizures in infancy, cerebellar ataxia, and hearing loss. Moreover, magnetic resonance imaging evidenced subtle symmetrical signal changes in the cerebellar dentate nuclei (96).
fxyd2. Gene linkage studies identified the FXYD domain containing ion transport regulator 2 (FXYD2) mutations in a family with dominant isolated renal Mg2+ wasting (IDH; OMIM 154020) (339). The patients in this family presented with low serum Mg2+ values (±0.4 mM), while other plasma electrolytes, including Na+, K+, Ca2+, CU, and HCO7, were normal. Urinary Mg2+ excretion was increased, whereas Ca2+ excretion was slightly lowered (172). The c.G121A mutation results in a p.G41R missense mutation at the protein level, which causes misrouting of FXYD2 to the membrane (66). FXYD2 encodes for the •y-subunit of the Na+-K+-ATPase. Although the exact role of FXYD2 in the DCT is unknown, it has been hypothesized to stabilize the Na+-K+-ATPase, influencing the membrane potential necessary for Mg2+ transport (345). However, other reports suggest that it may function independently as an inward rectifier channel (464). Functional analysis of the patient’s proximal tubular cells showed no differences in Na+, K+, or ATP affinity of the Na+-K+-ATPase, but demonstrated a lower FXYD2 protein expression (67).
hnf1b. Renal cysts and diabetes syndrome (RCAD; OMIM 137920) is caused by mutations in hepatocyte nuclear factor 1 (HNF1) and consists of a heterogeneous group of symptoms including renal cysts (±70% of patients), maturity onset diabetes of the young subtype 5 (MODY5; ±50%), and hypomagnesemia (±45%) (11, 82). HNF1 is a transcription factor regulating gene expression in kidney development (331). FXYD2b expression is one of several genes that is regulated by HNF1 (155), which may explain its role in renal Mg2+ handling. However, the possibility that HNF1 regulates other DCT genes involved in renal Mg2+ transport cannot be excluded.
pcbd1. In a small cohort of three patients, mutations in pterin-4 a-carbinolamine dehydratase 1 (PCBD1) have been linked to hypomagnesemia, renal Mg2+ wasting, and MODY5-like diabetes (154). PCBD1 mutations are known to cause transient neonatal hyperphenyalaninemia and high urinary levels of primapterin (HPABH4D; OMIM 264070) (518, 519). HPABH4D patients are diagnosed at birth by Guthrie testing and suffer from a transient, benign defect in impaired BH4 regeneration. A follow-up study of three patients at ± 18 yr of age showed that the patients display a mild hypomagnesemia (±0.6 mM) and MODY5-like diabetes, but no renal cysts (154). Interestingly, serum and urinary Na+, K+, Ca2+, and CU levels were within the normal range. The phenotype of the HPABH4D patients resembles that of RCAD patients, and the treatment regime consists of sulfonylureas and Mg2+ supplements. The origin of renal cysts in RCAD patients may be traced to the CD where HNF1 regulates PKHD1 (194). However, since PCBD1 is not expressed in CD, HPABH4D patients are protected from cyst formation (154).
slc12a3. Hypomagnesemia and hypokalemia are the cardinal symptoms of a hereditary electrolyte disorder characterized by Dr. Gitelman in 1969 that has been known since as Gitelman’s syndrome (175). Patients present with tetany, paresthesias, and chondrocalcinosis (284). The severity of the symptoms depends on the degree of hypokalemia. Except hypokalemia and hypomagnesemia, laboratory investigations often show metabolic alkalosis and hypocalciuria, sometimes associated with a mild hypotension and prolonged QT interval. SLC12A3 encodes the thiazide-sensitive Na+-Cl~ cotransporter (NCC), and mutations here cause Gitelman’s syndrome (165, 481). Patients with Gitelman’s syndrome are often treated with oral Mg2+ supplements (284). Interestingly, in some patients Mg2+ supplementation restores normal K+ levels, suggesting that hypokalemia is secondary to hypomagnesemia (205). This hypothesis is further substantiated by the NCC KO mouse, which is hypomagnesemic but does not display K+ disturbances under basal conditions (460). NCC KO mice have markedly reduced TRPM6 expression levels, possibly explaining the renal Mg2+ wasting observed in Gitelman’s syndrome (368). However, the mechanism by which a loss of NCC function results in reduced TRPM6 expression remains unresolved. It has been suggested that the atrophy of the DCT segment observed in KO mice may partially explain this phenomenon (321).
l) slc12a1, bsnd, clcnkb, and kcnjI. Bartter’s syndrome was originally described by Dr. Bartter in 1962 and is characterized by salt wasting, hypokalemic alkalosis, elevated plasma renin and aldosterone levels, and low blood pressure (40). Mutations in SLC12A1, encoding NKCC2, Barttin, ClC-Kb, KCNJ1, encoding ROMK, or CaSR form the genetic basis of Bartter’s syndrome (50, 477-479). Mild hypomagnesemia is sometimes observed in Bartter’s patients, which may be explained by a reduced driving force for paracellular Mg2+ reabsorption in the TAL. Compensation for reduced TAL Mg2+ reabsorption may take place in the DCT, which explains why Bartter’s patients often have normal Mg2+ levels. ClC-Kb and Barttin are also expressed in DCT, which justifies why patients with mutations in these genes more often show hypomagnesemia (261).
Drug-induced hypomagnesemia
Diuretics.
Hypomagnesemia has been associated with diuretics targeting the TAL and DCT segments of the kidney. In 1968, Duarte (127) reported renal Ca2+ and Mg2+ wasting as a consequence of furosemide treatment. Furosemide inhibits the activity of NKCC2, reducing the positive transepithelial membrane potential that drives paracellular Mg2+ transport in TAL (408). Although the incidence of furosemide-induced hypomagnesemia is unclear, a significant number of patients who use it may suffer from Mg2+ wasting (92, 330). In a recent animal study, furosemide treatment did not result in hypomagnesemia since increased TRPM6 expression in the DCT was able to compensate for reduced Mg2+ reabsorption in TAL (530). The clinical effects of furosemide treatment on Mg2+ levels may, therefore, depend on the individual’s ability to compensate at DCT level.
The use of thiazide diuretics, which target NCC in DCT, frequently induces renal Mg2+ wasting (366). Although most studies do not report hypomagnesemia in patients being treated with thiazide diuretics, some patient groups may be at risk (101). Patients with low initial Mg2+ values, such as elderly patients or patients with chronic heart failure, may develop hypomagnesemia after chronic thiazide treatment (238, 285). In mice, thiazide treatment reduces the renal expression of TRPM6, explaining the high urinary Mg2+ excretion (368). However, in most patients these effects may be small and the clinical consequences may depend on the patients’ basal serum Mg2+ values.
egfr inhibitors.
In 2005, it was first reported that the use of the EGFR inhibitor cetuximab can result in severe hypomagnesemia (459). Cetuximab (erbitux) is a monoclonal antibody against the EGFR and is generally prescribed for the treatment of colorectal or head and neck cancer. up to 50-60% of patients using cetuximab may develop, and 10-20% display serum levels below 0.4 mM (341, 510). A recent meta-analysis showed a RR of 3.87 (396). Immediately after the first reports of cetuximab-induced hypomagnesemia, questions were raised whether the EGFR inhibitor erlotinib (tarceva) may have similar effects (16). However, as of this writing, no clinical reports on the Mg2+ status after the use of erlotinib are available. Animal studies demonstrated a small reduction in serum Mg2+ values after erlotinib administration (122). Erlotinib is generally administered as a tablet that also contains Mg2+ stereate. The copresence of Mg2+ in these tablets may explain the absence of clinical consequences of erlotinib administration on serum Mg2+ levels.
proton pump inhibitors.
In 2006, the use of protein pump inhibitors (PPI) was associated with hypomagnesemia for the first time in two separate patients receiving long-term omeprazole treatment (144). Since then, many new cases of ppI-induced hypomagnesemia have been reported (295). A recent systematic review of 36 cases demonstrated that discontinuation of ppIs resulted in recovery from hypomagnesemia within 4 days, and rechallenge led to reoccurrence within 4 days (226). Urinary Mg2+ excretion is low in these patients, suggesting normal kidney function and thus an effect of PPI use on intestinal Mg2+ absorption. In mice, administration of omeprazole increased the expression of TRPM6 in colon (300). Therefore, it was hypothesized that omeprazole may inhibit the activity of the colonic H+-K+- ATpase, resulting in reduced extrusion of protons into the colon. Since TRPM6 activity increases at lower external pH, decreased proton secretion may reduce TRPM6 activity, for which increased TRPM6 expression may compensate (300, 313). However, increased TRPM6 expression may not be sufficient to prevent malabsorption of Mg2+ in colon in all patients. Individual variability of this compensatory mechanism may explain why only a subset of ppI users develop hypomagnesemia.
calcineurin inhibitors.
The calcineurin inhibitors (CNI) cyclosporin A (CsA) and tacrolimus (FK506) are currently the first immunosuppressant drugs of choice after transplantation. The use of CNIs has been associated with hypertension and renal Mg2+ wasting (39, 517). Whereas the hypertension may be explained by an increased activity of NCC, renal Mg2+ wasting is not fully understood (242). Up to 90% of all patients suffer from significantly reduced serum Mg2+ levels after initiation of CsA treatment, and in a recent cohort even 35% of patients remained hypomagnesemic despite Mg2+ supplementation (39, 439, 517). In rats, CsA and FK506 treatment increased renal Mg2+ wasting and hypomagnesemia (29, 365). In FK506-treated rats TRPM6 mRNA is downregulated, resulting in dramatically increased Mg2+ excretion (365). In a recent study with CsA treatment in rats, TRPM6, TRPM7, and EGF mRNA expression were reduced, although the fractional excretion of Mg2+ did not significantly change (306). Interestingly, EGF treatment did not change Mg2+ excretion and TRPM6 expression in CsA-treated rats, whereas it reduced Mg2+ wasting and increased TRPM6 expression in control rats. These results suggest that CsA may interfere with the EGF signaling pathway in DCT cells. Patients receiving CNI treatment are generally supplemented with Mg2+ to prevent hypomagnesemia.
cisplatin/carboplatin.
Already from the introduction of cisplatin (cis-diamminedichloridoplatinum) as anti-cancer therapeutic, hypomagnesemia has been reported in ~40%- 80% of treated patients (453, 593). Nephrotoxicity is a common side effect of cisplatin treatment, mainly as a consequence of proximal tubule cisplatin accumulation, which results in necrosis of the tubular cells (298). However, the effect of cisplatin on electrolyte wasting is highly specific for Mg2+; concomitant Ca2+ and K+ wasting is only observed in severely hypomagnesemic patients. This suggests that hypomagnesemia cannot be explained by the nephrotoxictiy and that Ca2+ and K+ disturbances are secondary to Mg2+ wasting. Treatment with carboplatin [paraplatin, cisdiammine(1,1-cyclobutanedicarboxylato)platinum], another platinum derivate, results in similar side effects including hypomagnesemia (142, 495). Recently, two animal studies have examined the effects of cisplatin treatment in detail (305, 529). Both observe significant downregulation of TRPM6 mRNA levels, although the causative mechanisms of reduced TRPM6 expression may differ. In the mice study of Van Angelen et al. (529), all DCT markers including parvalbumin and NCC are reduced, suggesting that cisplatin treatment induced atrophy of the DCT segment. In rats, Ledeganck et al. (305) demonstrated no effects on NCC expression, which implies that the DCT cells are still intact. Nevertheless, TRPM6 and EGF expression were reduced. Both studies show compensation of Mg2+ uptake in the TAL by increased expression of claudins. Generally, patients that develop hypomagnesemia during cisplatin treatment are supplemented by adding Mg2+ to pre- and posthydration fluids to prevent hypomagnesemia.
Antimicrobials.
Although several classes of antimicrobials may cause hypomagnesemia, the underlying mechanisms leading to Mg2+ wasting differ greatly. Aminoglycoside antibiotics (AGA) including gentamycin, neomycin, tobramycin, and amikacin may induce renal Mg2+ wasting (562, 585). Estimates of the incidence of hypomagnesemia as a consequence of AGA use range from 20 to 80% (157, 562). AGAs may activate the CaSR, resulting in reduced paracellular Mg2+ transport in the TAL and inhibition of Mg2+ transport in the DCT. Studies in MDCT cells show reduced PTH-activated Mg2+ transport (269). Moreover, animal studies have evidenced that use of AGAs cause hypomagnesemia, due to reduced expression of NKCC2 that provides the driving force for TAL Mg2+ transport (167, 446). In a recent study with gentamycin-treated rats, TRPM6 expression was upregulated, suggesting that the DCT compensates for reduced TAL Mg2+ reabsorption (307).
Pentamidine is an antimicrobial against Pneumocystis ji- rovecii infections that are often diagnosed in AIDS patients. The use of pentamidine has been associated with severe hypomagnesemia due to renal Mg2+ wasting at the start of the 1990s (65, 188, 465). The exact mechanism of reduced Mg2+ reabsorption remains unresolved. However, pentamidine reduces ENaC activity, resulting in hyperkalemia (283). Moreover, there have been reports of tubular necrosis after pentamidine treatment (546), which may cause atrophy of the DCT segment.
Rapamycin (sirolimus) is an antibiotic that is frequently used to prevent organ rejection after transplantation. Rapamycin inhibits mToR activity, and its use has been associated with hypomagnesemia in 10-25% of patients (23, 535). Rapamycin-treated rats exhibit reduced expression of NKCC2 (100). Interestingly, TRPM6 expression was increased in the same study. This could be compensation for the reduced Mg2+ uptake in TAL, but a direct effect of rapamycin on TRPM6 expression cannot be excluded. A recent in vitro study showed the opposite effect; rapamycin decreased TRPM6 expression in an EGF-dependent manner (255).
Amphotericin B is an antifungal agent that has been associated with hypomagnesemia and hypokalemia (38). The mechanism underlying urinary Mg2+ wasting in these patients is unknown. Oral Mg2+ supplementation together with amiloride treatment is generally used to restore Mg2 + levels (178).
Foscarnet inhibits viral DNA polymerases by chelating divalent cations and, therefore, its use may cause hypomagnesemia (169). Patients also suffer from hypocalcemia and hypokalemia, which may be secondary to the Mg2+ disturbances (253, 369). Until now, no studies have examined the effect of forscarnet on the expression of renal ion transporters. It would be interesting to examine whether forscarnet exhibits effects beyond its chelating function.
CONCLUDING REMARKS
Over the last decade, Mg2+ has been considered as a treatment for several major diseases including preeclampsia, stroke, myocardial infarction, and asthma in several large- scale clinical trials. These findings have raised interest in Mg2+ among neurologists, cardiologists, and pneumologists. Nevertheless, Mg2+ levels are still not determined routinely in daily clinical practice, eventhough up to 60% of all critically ill patients are Mg2+ deficient (84, 145). Serum Mg2+ should be determined standardly, alongside Na + , K + , and Ca2+ measurements in patients. Mg2+ disturbances can cause muscle cramps, arrhythmias, and seizures, and therefore, Mg2+ should be considered when patients present in clinic with these symptoms. However, the molecular mechanisms underlying the effects of Mg2+ in brain, heart, and lung are still largely unknown. Finding the molecular basis of the role of Mg2+ in these diseases may further extend the significance of Mg2 + therapies in clinic.
Genetic and drug-induced disorders of Mg2+ homeostasis have enhanced the knowledge on Mg2+ (re)absorption in the kidney and intestine. These studies form a perfect example of the powerful interaction of clinical and fundamental studies. For instance, the increased knowledge on the role of EGF in renal Mg2+ handling has resulted in the standardization of Mg2+ measurements in patients using EGFR blockers. This resulted in early detection of Mg2+ disturbances and changes in treatment strategies. Through synergistic clinical and fundamental efforts in the fields of brain, heart, and lung Mg2+ research, the unexplained role of Mg2+ in, among others, migraine, depression, epilepsy, COPD, and hypertension, may be elucidated. Over the last decades Mg2+ research has been centered around the kidney and the intestine. By extending the field to the heart, brain, and lungs and by involving both fundamental and clinical researchers, Mg2+ will never be thought of as “a forgotten cation” anymore.
ACKNOWLEDGMENTS:> We acknowledge the careful reading of the manuscript by Dr. Anke Lameris, Dr. Jenny van der Wijst, Cas van der Made, and Judy Lin. Robert Boensma drew the illustrations.
Address for reprint requests and other correspondence: R. J. M. Bindels, Radboud University Medical Center, Dept. of Physiology 286, PO Box 9101,6500 HB Nijmegen, The Netherlands (e-mail: Rene.Bindels@radboudumc.nl).
GRANTS: This work was supported by grants from the Netherlands Organization for Scientific Research [NWO Vici Grant (to J. G. J. Hoenderop), 016.130.668, and ZonMw 9120.8026], Dutch Kidney Foundation (CP10.11, IP10.27, IP11.46, and PhD12.14), and EURenOmics funding from the European Union Seventh Framework Program (FP7/ 2007-2013, agreement no. 305608).
DISCLOSURES: No conflicts of interest, financial or otherwise, are declared by the authors.
REFERENCES are in PDF
short url = is.gd/Mg2015