How Vitamin D prevents many cancers (c-MYC)
Vitamin D receptor as a master regulator of the c-MYC/MXD1 network
PNAS November 13, 2012 vol. 109 no. 46 18827-18832
Reyhaneh Salehi-Tabara, Loan Nguyen-Yamamotoa, Luz E. Tavera-Mendozaa, Thomas Quailb, Vassil Dimitrovb, Beum-Soo Anb,c, Leon Glassb, David Goltzmana,b, and John H. Whitea,b, john.white@mcgill.ca
A Departments of Medicine and
B Physiology, McGill University, Montreal, QC, Canada H3G 1Y6; and
C Department of Biomaterial Science, College of Natural Resources and Life Science, Pusan National University, Republic of Korea
Edited* by Hector F. DeLuca, University of Wisconsin-Madison, Madison, WI, and approved October 2, 2012 (received for review June 12, 2012)
Vitamin D signaling regulates cell proliferation and differentiation, and epidemiological data suggest that it functions as a cancer chemopreventive agent, although the underlying mechanisms are poorly understood. Vitamin D signaling can suppress expression of genes regulated by c-MYC, a transcription factor that controls epidermal differentiation and cell proliferation and whose activity is frequently elevated in cancer. We show through cell- and animal-based studies and mathematical modeling that hormonal 1,25-dihydroxyvitamin D (1,25D) and the vitamin D receptor (VDR) profoundly alter, through multiple mechanisms, the balance in function of c-MYC and its antagonist the transcriptional repressor MAD1/MXD1. 1,25D inhibited transcription of c-MYC–regulated genes in vitro, and topical 1,25D suppressed expression of c-MYC and its target setd8 in mouse skin, whereas MXD1 levels increased. 1,25D inhibited MYC gene expression and accelerated its protein turnover. In contrast, it enhanced MXD1 expression and stability, dramatically altering ratios of DNA-bound c-MYC and MXD1. Remarkably, F-box protein FBW7, an E3-ubiquitin ligase, controlled stability of both arms of the c-MYC/MXD1 push–pull network, and FBW7 ablation attenuated 1,25D regulation of c-MYC and MXD1 turnover. Additionally, c-MYC expression increased upon VDR knockdown, an effect abrogated by ablation of MYC regulator β-catenin. c-MYC levels were widely elevated in vdr−/− mice, including in intestinal epithelium, where hyperproliferation has been reported, and in skin epithelia, where phenotypes of VDR-deficient mice and those overexpressing epidermal c-MYC are similar. Thus, 1,25D and the VDR regulate the c-MYC/MXD1 network to suppress c-MYC function, providing a molecular basis for cancer preventive actions of vitamin D
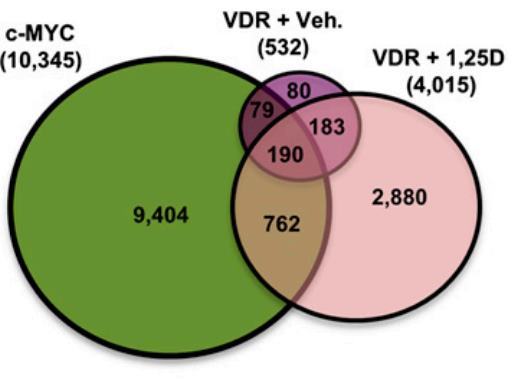
Results of a comparative analysis of overlap of genomic binding sites from ChIP sequencing studies of c-MYC and the VDR (from cells treated with vehicle [Veh] or 100 nM 1,25D for 36 h) performed in related lymphoblastoid cell lines.
PDF of paper and appendix are both attached at the bottom of this page
Interview of Dr. White
by Dr. Passwater, WholeFoods Magazine. March 2013
Vitamin D slows the progression of cells from premalignant to malignant states, keeping their proliferation in check. A team of researchers at McGill University has discovered a molecular basis for the potential cancer-preventive effects of vitamin D (1). The team, led by McGill professors John White and David Goltzman, of the Faculty of Medicine’s department of physiology, discovered that the active form of vitamin D acts through several mechanisms to inhibit both the production and function of a protein that drives cell division and is active at elevated levels in more than half of all cancers.
The initial observation that vitamin D deficiency increases the risk of many cancers, especially breast and colon cancers, was made in the 1970s by brother epidemiologists, Cedric Garland, Ph.D., and Frank Garland, Ph.D. (2). When I wrote my book on cancer and nutrition in 1978, I included five of the mortality maps from National Cancer Institute’s Atlas of Cancer Mortality for U. S. Counties from 1950–1969 to illustrate the epidemiological evidence observed by the Drs. Garland about sunlight exposure (latitude) and cancer incidence.
Vitamin D was thought of only in terms of bone health for decades. It was realized that vitamin D was not a classical vitamin because classical vitamins are part of specific coenzyme systems. Vitamin E is another example of a nonclassical vitamin in that it can act as a general antioxidant. Vitamin D, when converted in the body to its biologically active form, has hormone-like actions and became recognized as a hormone. Vitamin D has become very sexy in the last five years because it has been found to be involved in many biological pathways and a deficiency increases the risk of many diseases. Much of this research stems from the findings of John H. White , Ph.D., as he has elucidated how vitamin D interacts with more than 1,000 genes.
Professor White , who obtained his Ph.D. in 1987 from Harvard, is a molecular biologist and molecular geneticist who has made numerous broad-ranging contributions to the fields of gene regulation and vitamin D physiology. His work on the molecular mechanisms of vitamin D action opened up the field of study of vitamin D as an inducer of antimicrobial innate immunity in humans, and has provided fundamental insights into its potential role as a cancer chemopreventive agent. Dr. White is a professor in the departments of physiology and medicine at McGill University in Montreal, Canada.
Passwater : Dr. White , congratulations on your recent article published in the Proceedings of the National Academy of Sciences explaining how vitamin D works to regulate cell division (1). This is an extremely important research finding. Why did you become a molecular geneticist in the first place? What drew you to molecular genetics?
White : Molecular biology introduced me to molecular genetics. I became interested in molecular biology as a Ph.D. student. Molecular biology is not a scientific discipline like biochemistry or physiology, but rather a series of elegant molecular techniques for studying biochemistry, physiology and other related fields. Many of these techniques are gene-based; so-called “recombinant DNA technology” and “gene therapy” are components of the molecular biology arsenal, for example.
Passwater : Why did you become interested in gene regulation?
White : Molecular biology introduced me to genes and how they are regulated. As you know, genes encode the information necessary to build proteins. For non-scientist readers to understand what that means, one has to take a step back and understand what cells are.
Cells are units of life, from simple unicellular organisms to complex multicellular ones like humans. It is helpful to imagine that a cell is like a city or city-state. While proteins contribute to the infrastructure of the cell, they are above all like the specialized individuals who make the city work, with all of the specialized tasks that entails. Some of these jobs (e.g., police, bus drivers) are common to all cities, while certain types of expertise (e.g., specialized types of engineers or scientists) are found only in selected areas.
Similarly, cells in the liver or, say, the brain will produce common proteins while others will be specific to each cell type. Thus, protein production is specific to the function of a given cell to some degree.
All of this means that protein production must be carefully regulated. The first and, I believe, most important point of this regulation is the control of the first step: gene transcription. Although it is not evident at this point of the conversation, this is highly relevant to how vitamin D works at the molecular level.
Passwater : That’s a nice analogy. How did vitamin D enter your research picture? Were you aware of other research on vitamin D and cancer first, or was it your awareness of vitamin D and genes that came first?
White : The awareness of vitamin D and genes came first, although it took me a while to get there. When I finished my Ph.D., I went to work in a lab that studied how estrogen worked—but not physiologically. The lab had no particular interest in female reproductive function, for example, but rather in the nitty-gritty details of how estrogen worked at the molecular level.
Many readers are aware that estrogen is a steroid hormone. Steroid hormones are small molecules that can freely enter cells. Estrogen acts by binding to a specific protein called the estrogen receptor, which is found in an inner compartment of the cell called the nucleus. The nucleus contains the cell’s DNA (i.e., its genes). Steroid hormone receptors function as “gene switches” and regulate the process, gene transcription, mentioned previously. Estrogen physiology is thus manifested by estrogen’s control of the production of specific proteins through gene transcription in specific cells that are necessary for the menstrual cycle, bone health and more.
Similarly, the active form of vitamin D (commonly called 1,25-dihydroxyvitamin D or calcitriol) binds to a specific receptor, the vitamin D receptor, which regulates the transcription of specific genes. The active form of vitamin D is four metabolic steps removed from cholesterol , another well-known, but non-hormonal steroid. The physiological effects of vitamin D thus arise through the regulation of gene transcription. Vitamin D was discovered for its capacity to regulate the body’s uptake and use of calcium, in particular its role in maintaining normal bone health.
My interest in vitamin D was piqued in the late 1990s by a colleague who mentioned in passing that the application of vitamin D to cancer cells grown in tissue culture arrested their division. This so-called “non-classical” action of vitamin D became a main focus of my lab because I realized that I could apply my expertise in studying how estrogen worked to the study of vitamin D.
Passwater: You have identified about 1,000 genes regulated by vitamin D . That is an absolutely amazing feat that has changed our understanding of vitamin D and health. Was the effect of vitamin D on genes known before you identified them? Was vitamin D still looked upon in classical vitamin terms when you started your vitamin D/gene research?
White : In the late 1990s, there was increasing awareness of the non-classical actions of vitamin D and accumulating population-based studies linking vitamin D deficiency with increased rates of specific types of cancers. However, the molecular details of these actions were definitely lacking. I think it is fair to say that my lab was the first to take a comprehensive approach to try and understand these actions by determining on as large a scale as possible the identity of the genes regulated by vitamin D. Before that point, we were aware of only a handful of genes under the control of vitamin D.
Passwater : What was your first important finding in this aspect of your research?
White : That’s a difficult question to answer. Perhaps, in retrospect, the first important finding was what didn’t happen, that vitamin D and the vitamin D receptor were not strongly stimulating the production of a protein that acted as a brake on cell division. We saw lots of changes in gene transcription that were consistent with cell division being arrested. However, the vast majority of these corresponded to the loss of production of proteins necessary for cell division. Moreover, and this is important, we couldn’t tell which changes were a product of the arrest in cell division and which ones were driving it. It took us seven or eight years to begin to figure it out.
As an aside, I should point out that one of the early products of our approach of large-scale identification of vitamin D “target” genes was the realization that vitamin D also controlled the function of the immune system and its primary responses to infection (3). This is one of the fortunate and serendipitous aspects of research, and led to another major research effort in the lab. However, as I said before, the control of cell division by vitamin D was a tougher nut to crack.
Passwater : Wow! The mechanism by which vitamin D regulates the primary response to infection is another outstanding discovery. But, don’t let me interrupt you, where did you go from there regarding elucidating the mechanism for vitamin D regulation of cell division?
White : We came to realize that many genes whose transcription was reduced in vitamin D-treated cells were also regulated by a protein called cMYC. cMYC is at the same time similar to the vitamin D receptor and in some ways the opposite of it . It is similar in that, like the receptor, cMYC is a regulator of specific genes’ transcription. However, it is the opposite in the sense that, while vitamin D working through its receptor acts to block cell division, cMYC acts to stimulate it. Not surprisingly, the function of cMYC is elevated in about 50% of all cancers, which are of course diseases of uncontrolled cell division.
Slowly, in retrospect perhaps too slowly, it dawned on us that the vitamin D receptor was acting by directly controlling cMYC function . What came as such a surprise, and I think that this is why our paper is important, is that vitamin D and the vitamin D receptor control cMYC function by multiple mechanisms.
They depress the rate of transcription of the gene encoding cMYC and thus the production of new cMYC protein.
In addition, they increase the rate of degradation, or turnover, of existing cMYC protein. *Moreover, they have opposite effects on the production and turnover of a protein called MXD1 that antagonizes cMYC function, leading to dramatic swings in the relative amounts of cMYC and MXD1 in vitamin D-treated cells.
This is why we called the vitamin D receptor a “master regulator” of cMYC function in the title of the paper.
Passwater: Is this like putting a brake on runaway cell division or cancer?
White : We think that vitamin D’s control of cMYC function is one of the key elements of its capacity to control cell division and we believe that it represents an important aspect of its anticancer properties. I should point out by “anticancer” I mean the prevention of cancer development , not the treatment of existing cancers. As you know, as cancers progress they can become resistant to various types of therapeutic approaches, and vitamin D treatment is no exception. One day, we would like to understand the mechanisms of resistance to vitamin D of late-stage cancers.
Passwater : Yes, I agree that cancer cells are a different beast and develop their own defense mechanisms. I find a recently published article particularly disturbing (4). The researchers suggest that only one in 10,000 tumor cells may be responsible for keeping a cancer growing. Your research focuses on cancer prevention, however, it hasn’t been shown that controlling cMYC regulation will not put a brake on some cancers. Nor has it been shown that vitamin D will not affect other vitamin D-regulated mechanisms involved in the multi-step DINOMIT theory of cancer.
Vitamin D is incredibly inexpensive, safe and doesn’t interfere with any cancer treatment. I would like to see your findings about cancer prevention tested in cancer patients as well as studied for the prevention of cancer. In the meantime, your findings do suggest that if people maintain adequate levels of vitamin D, their risks of many cancers can be reduced. If people don’t develop cancer in the first place, the need for destroying cancer cells becomes moot.
I have been following the tumor suppressor protein p53 and cancer for about 30 years. Does cMYC fit into the p53 regulatory mechanism?
White : It’s not something that we have been looking at. However, there is some evidence linking elevated transcription of the gene encoding cMYC to chromosomal instability resulting from mutations in p53.
Passwater : Getting back to your findings, how are vitamin D and vitamin A involved in co-regulating genes?
White: The active form of vitamin A in most tissues is retinoic acid. Retinoic acid binds to a receptor related to the vitamin D receptor and thus also functions as a “gene switch.” Like vitamin D, retinoic acid can also regulate cell division. That being said, the extent to which vitamin D and retinoic acid can coregulate gene transcription has not been studied in great detail, but is probably rather limited.
Passwater : We often discuss the relationship between dietary vitamin D (cholecalciferol) and the form of vitamin D that circulates in the blood (25-hydroxy vitamin D or calcidiol) in this column. What blood levels of vitamin D should people have to optimize cMYC regulation?
White : We didn’t look at the levels of vitamin D in the blood in the animal studies. The major circulating form of vitamin D in animals and humans (commonly called 25-hydroxy vitamin D) is a precursor of the active form (1,25-dihydroxyvitamin D). We treated the skin of normal mice with pharmacological concentrations of the active form of vitamin D and saw that levels of cMYC were reduced, effectively reproducing what we saw with human cells propagated in tissue culture. Otherwise, we compared the levels of cMYC in normal mice and mice that could not produce vitamin D receptors, in other words in mice where vitamin D could not function. We saw elevated cMYC production in the skin and colon of “vitamin D defective” mice.
Passwater : How are your findings being received by the scientific community?
White : The results were presented at a workshop on vitamin D function held in Houston last summer and were well received. The paper was also voted one of the top 10 research achievements of 2012 by the Canadian Cancer Society.
Passwater : Where does your research go from here? What are your other research interests in your lab?
White : We are pursuing our vitamin D work on two fronts. We are trying to determine how vitamin D regulates cMYC stability. This may be important, as its stability is controlled by mechanisms that also regulate the stability of a host of other proteins that, like cMYC, act to drive cells through cell division; cMYC in other words may represent the tip of an iceberg in terms of vitamin D action. We are also continuing our work on how vitamin D stimulates cellular responses to bacterial infection.
Passwater : Dr. White , thank you for your discoveries and for chatting with us about their meaning. WF
Dr. Richard Passwater is the author of more than 40 books and 500 articles on nutrition. Dr. Passwater has been WholeFoods Magazine’s science editor and author of this column since 1985. More information is available on his Web site, www.drPasswater.com.
References
R. Salehi-Tabar et al., “Vitamin D Receptor as a Master Regulator of the C-MYC/MXD1 Network,” Proc. Nat. Acad. Sci. USA 109 (46), 18827–18832 (2012).
C.F. Garland and F.C. Garland, “Do Sunlight and Vitamin D Reduce the Likelihood of Colon Cancer?” Int. J. Epidemiol. (3), 227–231 (1980).
T.T. Wang et al., “Cutting Edge: 1,25-dihydroxyvitamin D3 Is a Direct Inducer of Antimicrobial Peptide Gene Expression,” J. Immunol. 173,2909–2912 (2012).
A. Kreso et al., “Variable Clonal Repopulation Dynamics Influence Chemotherapy Response in Colorectal Cancer,” Science Dec. 13, 2012.
See also VitaminDWiki
See also web
Finding cancer's weakest link. Dec 2011 full free text on-line
MYC and metastasis March 2011 full free text on-line
NEWLY DISCOVERED EFFECTS OF VITAMIN D ON CANCER Science Blog on this study Nov 2012
UVA causes skin cancer, perhaps UVB (Vitamin D) prevents skin cancer – Jan 2017