Trichloroethylene increases risk of Parkinson's by 5X - Vitamin D decreases the risk
The majority of this page was written by Claude AI April 2026
Solvent in the water, disease in the brain: TCE, Parkinson's and autoimmunity
Trichloroethylene (TCE) is now among the most strongly supported environmental causes of Parkinson's disease and a well-established trigger of several autoimmune disorders, most prominently systemic sclerosis (scleroderma). A landmark 2023 JAMA Neurology study of ~340,000 Marines at Camp Lejeune found a 70% higher risk of Parkinson's disease in those exposed to contaminated drinking water, and a 2025 nationwide Medicare analysis of 1.35 million older Americans extended the signal to ambient air exposure. In parallel, meta-analyses show roughly a doubling of scleroderma risk with TCE exposure, particularly in men, while Chinese occupational cohorts have described a severe, HLA-B*13:01–linked TCE hypersensitivity syndrome with 27-fold higher odds in carriers. After decades of voluntary controls, EPA finalized a near-total ban on TCE under TSCA in December 2024 — though that rule is now tangled in 2025–2026 litigation and partial postponements. Vitamin D is separately protective against both dopaminergic injury and autoimmune incidence, but direct vitamin D × TCE interaction studies are essentially absent; any claim of a protective effect remains a biologically plausible hypothesis, not an evidence-based conclusion.
Camp Lejeune and the epidemiology of TCE-induced Parkinson's
The case that TCE causes Parkinson's disease (PD) rests on a converging set of studies with remarkably consistent direction. Goldman et al. (JAMA Neurology 2023;80:673–681) compared 172,128 Marines stationed at Camp Lejeune (1975–1985, when monthly median TCE in drinking water was 366 µg/L — over 70× the EPA Maximum Contaminant Level of 5 µg/L) with 168,361 Marines at uncontaminated Camp Pendleton. After a mean ~34-year latency, PD prevalence was 0.33% vs 0.21%, yielding an adjusted OR 1.70 (95% CI 1.39–2.07), rising to 1.81 (1.46–2.24) for probable PD and 1.82 (1.41–2.36) in patients seen in the VHA before diagnosis. Prodromal features (tremor, anxiety, erectile dysfunction) were also elevated 8–19% in exposed Marines without diagnosed PD, and a follow-up Goldman et al. paper (Movement Disorders 2024;39:1732–1739) reported faster progression to falls (HR 2.64), psychosis (HR 2.19) and fractures (HR 2.44) in exposed PD patients, with a significant dose-response for falls (p-trend 0.032).
The twin study by Goldman et al. (Annals of Neurology 2012;71:776–784) remains the cleanest case-control estimate, exploiting 99 twin pairs discordant for PD from the NAS/NRC WWII veteran cohort. After blinded expert rating of occupational and hobby exposures, ever-use of TCE yielded OR 6.1 (95% CI 1.2–33), rising to OR 8.9 (1.7–47) for ≥6 months' exposure and OR 10.5 (0.97–113) for perchloroethylene (PCE). A 2025 Medicare nationwide case-control study by Krzyzanowski, Racette and colleagues (Neurology 105:e214174) found a dose-dependent RR 1.10 for highest-vs-lowest decile of ambient TCE across 221,789 incident PD cases and 1.13 million controls — modest per-person risk but enormous population attributable impact given how widespread low-level air exposure is. An accompanying editorial by Dorsey and Tanner called the finding confirmatory of the Camp Lejeune signal. The civilian Camp Lejeune mortality cohort (Bove et al., Environmental Health 2014) had produced a directionally consistent but imprecise PD mortality HR 3.13 (0.76–12.81), and the Gash et al. 2008 industrial cluster (Ann Neurol 63:184–192) documented overt parkinsonism in workers with 8–33 years of TCE contact. No formal meta-analysis yet exists because of heterogeneous exposure assessment, but the triangulation of a ~1.7-fold cohort signal, a ~6-fold occupational signal, and a 1.1-fold ambient-air signal constitutes a strong epidemiologic case.
The biology: mitochondria, α-synuclein and TaClo
TCE's mechanism reads like a laboratory recipe for Parkinson's pathology. Its lipophilic molecule readily crosses the blood-brain barrier, and one of its endogenously formed metabolites — 1-trichloromethyl-1,2,3,4-tetrahydro-β-carboline (TaClo), produced by Pictet-Spengler condensation of tryptamine with the TCE metabolite chloral — is a potent mitochondrial complex I inhibitor structurally analogous to MPTP/MPP+ and reportedly ~10× more potent. Rodent models consistently reproduce the PD phenotype: Gash et al. (2008) showed selective complex I impairment and nigrostriatal degeneration in F344 rats; Liu et al. (Neurotoxicology 2010) demonstrated dose-dependent dopaminergic neuron loss at 200/500/1,000 mg/kg with α-synuclein accumulation in the dorsal motor nucleus of vagus; Keane et al. (2019) documented a ~50% drop in tyrosine-hydroxylase-positive SNpc neurons after 8 weeks; De Miranda et al. (Neurobiology of Disease 2021) in aged Lewis rats linked TCE to elevated LRRK2 kinase activity, endolysosomal dysfunction, phospho-Ser129 α-synuclein accumulation and oxidative markers (4-HNE, 3-nitrotyrosine); and Adamson et al. (Toxicol Sci 2023) showed that inhalation at only 50 ppm (rats) or 100 ppm (mice) produced comparable or worse DA neuron loss than oral dosing, implicating vapor intrusion as the most worrying real-world route. Gut microbiome shifts mirroring idiopathic PD have also been reported (Ilieva et al. 2022). The pathology takes decades to manifest: mean latency in Camp Lejeune Marines was about 33.9 years from first exposure to diagnosis at age ~55, consistent with the long PD prodrome.
Positions of major organizations and the policy response
The VA has designated PD a presumptive service-connected disease for any veteran or reservist who served at Camp Lejeune for ≥30 days between August 1, 1953 and December 31, 1987 since January 13, 2017; the Camp Lejeune Justice Act (§804 of the PACT Act, signed August 10, 2022) created an exclusive cause of action in the US District Court for the Eastern District of North Carolina with a two-year filing window ending August 10, 2024. More than 500,000 administrative claims have been filed — one of the largest mass torts in US history — and by March 2026 DOJ had paid over $421 million under the Elective Option program, with cumulative offers of approximately $708 million across 2,531 settlements and ~3,700 cases still pending. The Michael J. Fox Foundation, coordinated by policy VP Ted Thompson, gathered 150,000+ petition signatures and submitted 90+ studies to EPA; the Parkinson's Foundation and NIH/NIEHS-funded laboratories (De Miranda at UAB, Greenamyre at Pittsburgh) align with this position. Dr. E. Ray Dorsey of the University of Rochester — author of Ending Parkinson's Disease (PublicAffairs, 2020) and the widely cited hypothesis paper "Trichloroethylene: An Invisible Cause of Parkinson's Disease?" (Journal of Parkinson's Disease 2023;13:203–218) — has argued in a 2024 J Parkinson's Dis position paper that Parkinson's is now "predominantly an environmental disease" driven by TCE, pesticides and air pollution.
The autoimmune signal: scleroderma leads, hypersensitivity is sharpest
Evidence for TCE as an autoimmune trigger is moderately strong for several conditions but strongest for systemic sclerosis. The Barragán-Martínez et al. meta-analysis (PLoS ONE 2012;7:e51506) pooled 33 studies and found organic solvent exposure associated with an overall autoimmune disease OR 1.54 (95% CI 1.25–1.92), with particular signals for systemic sclerosis, primary systemic vasculitis and multiple sclerosis. The solvent-specific meta-analysis by Zhao et al. (2016; 14 case-control studies, 1,657 cases/3,838 controls) pinned down a trichloroethylene–scleroderma OR of 2.07 (95% CI 1.34–3.17), alongside ketones (4.20), aromatic solvents (2.72) and halogenated solvents generally (1.49). The EPA/NIEHS review by Cooper et al. (Environ Health Perspect 2009;117:696–702) found a combined scleroderma OR of 2.5 (95% CI 1.1–5.4) in men but only 1.2 (0.58–2.6) in women occupationally exposed — a pattern reflecting occupational exposure opportunity, not biological susceptibility (women dominate TCE-induced autoimmunity when exposure is equalized). IARC classifies TCE as Group 1 (carcinogenic to humans); the NTP 15th Report on Carcinogens lists TCE as "known to be a human carcinogen"; and EPA's 2020 TSCA risk evaluation concluded TCE presents unreasonable risk including immunotoxicity in 52 of 54 conditions of use.
The most striking immune phenotype is occupational TCE hypersensitivity syndrome (OTHS), documented in Chinese electronics workers as a generalized exfoliative dermatitis with fever, hepatitis and lymphadenopathy (including Stevens-Johnson and toxic epidermal necrolysis) appearing 2–6 weeks after exposure onset. Li et al. (Environ Health Perspect 2007;115:1553–1556) reported an astonishing OR of 27.5 (95% CI 13.5–55.7) for HLA-B13:01 carriers among 121 cases vs 142 controls — one of the strongest pharmacogenomic associations ever described. A GWAS replication (174 cases, 1,761 controls) confirmed HLA-B13:01 as the dominant risk allele, with HHV-6 reactivation as a co-trigger. Beyond this, autoimmune hepatitis, systemic lupus erythematosus (signals in small case-control studies), primary systemic vasculitis and multiple sclerosis (Landtblom reviews) have been linked, while rheumatoid arthritis shows weaker and inconsistent associations.
Mechanisms of immune breakdown
TCE's immunotoxicity is mediated by reactive metabolites — trichloroacetaldehyde (chloral), trichloroethanol and dichloroacetic acid — that form covalent protein adducts (notably with albumin and liver proteins) acting as neoantigens recognized by CD4+ T cells. Work from Kathleen Gilbert's laboratory at UAMS in autoimmune-prone MRL+/+ mice (Khan 1995; Blossom 2012; Gilbert 2016 Epigenomics 8:633; Gilbert 2017; Byrum/Blossom 2019 Front Immunol) has shown that chronic low-dose TCE in drinking water expands effector/memory CD4+ T cells, skews them toward a Th1/IFN-γ phenotype, induces autoantibodies, and causes persistent DNA methylation changes at ~337,770 CpG sites — disproportionately at polycomb-binding sites and at the Ifng promoter — that remain after exposure cessation. A 2024 Toxicological Sciences paper (Choudhury, Byrum, Blossom Toxicol Sci 199:289–300) demonstrated that the oxidative metabolite trichloroacetaldehyde hydrate (TCAH) directly remodels DNA methylation in Th1-polarized CD4+ T cells, with effects "more robust in females than males" — a molecular correlate of the well-known female preponderance of autoimmunity. Phillips et al. (2019) provided human corroboration by showing occupational TCE exposure altered DNA methylation in peripheral blood, including hypomethylation of the IFN-γ regulator TRIM68. Oxidative stress — glutathione depletion, increased GSSG, mitochondrial ROS in the cerebellum — links this immunology to the neurotoxicity pathways above.
Sex differences. Women are disproportionately affected by idiopathic autoimmune disease generally (≈4:1 for SLE and scleroderma), and in TCE animal models female MRL+/+ mice show more pronounced CD4+ dysregulation and epigenetic changes than males. The hypothesized drivers are estrogen's amplification of Th1/Th17 responses, X-linked immune genes (TLR7, FOXP3, CD40L) that escape X-inactivation, and potentially sex-specific TCE metabolism. Occupational studies that appear to show male predominance reflect exposure, not susceptibility.
Who is exposed today, and how the regulatory picture changed
TCE remains ubiquitous despite declining use. It has been detected at approximately 852 Superfund sites — roughly 42% of all National Priorities List sites, second only to lead. US production fell from 220.5 million pounds (2012) to 171.9 million (2015), of which ~84% is an intermediate for the refrigerant HFC-134a and ~15% is solvent degreasing. Major historical exposure events, beyond Camp Lejeune (1953–1987; ~1 million people; TCE wells up to thousands of ppb, ~240–3,400× current safe limits), include the Woburn, Massachusetts childhood leukemia cluster (~21 cases 1969–1986; wells G and H contaminated at ≥41 ppb; basis of the W.R. Grace/Beatrice Foods litigation in Anderson v. Cryovac and Jonathan Harr's A Civil Action), the Hill AFB, Moffett Field, Tucson, Fort Ord, Lockformer and View-Master sites, and ongoing vapor-intrusion evacuations in New Jersey, Michigan, Ohio (tiered triggers at 2.1/6.3/20 µg/m³) and others. EPA's IRIS inhalation reference concentration is 2 µg/m³ (0.0004 ppm) — five orders of magnitude below the OSHA permissible exposure limit of 100 ppm and still far below NIOSH's REL of 25 ppm and ACGIH's TLV of 10 ppm.
The regulatory endgame finally arrived: on December 17, 2024 EPA published a final TSCA §6(a) rule (89 FR 102568) banning all manufacture, processing, distribution and use of TCE, with most uses prohibited by September 15, 2025; a small set of continued uses (battery separators, defense/aerospace parts cleaning, refrigerant manufacturing, nuclear fuel processing through 2028) operate only under Workplace Chemical Protection Plans with an interim Existing Chemical Exposure Limit of 0.2 ppm — 500× stricter than OSHA's PEL. However, the rule has been partially stayed: the Fifth Circuit granted a temporary stay January 13, 2025; the case moved to the Third Circuit as USW v. EPA No. 25-1055; the Trump administration delayed the effective date to March 21, 2025; and through successive postponements, the §6(g) exemption conditions are now pushed to May 18, 2026 (per Federal Register notice February 18, 2026), with EPA signaling reconsideration. Only Minnesota (2020, effective 2022) and New York (2020, effective 2021) had pre-empted this federally; the EU placed TCE on REACH Annex XIV with a 2016 sunset date. A companion December 18, 2024 rule restricted PCE.
Vitamin D: strong indirect plausibility, essentially no direct evidence
Vitamin D has robust evidence as an independent modifier of both PD and autoimmune risk, but no published study directly tests whether vitamin D status alters TCE-induced pathology in humans, and only a handful of animal studies touch adjacent antioxidant questions. On the Parkinson's side, vitamin D deficiency is highly prevalent in PD (often >50% of patients), VDR polymorphisms (FokI, BsmI) track with risk, and experimentally 1α,25-(OH)₂-vitamin D₃ protects mesencephalic dopaminergic neurons against glutamate and ROS toxicity (Ibi et al. 2001) and rescues 6-OHDA-lesioned rats (Lima et al. J Neuroinflammation 2018). The Suzuki et al. (Am J Clin Nutr 2013) RCT reported that 1,200 IU/day vitamin D₃ stabilized PD symptoms over 12 months in VDR FokI TT carriers. On the autoimmune side, the VITAL trial (Hahn et al. BMJ 2022;376:e066452), a 25,871-person RCT of 2,000 IU/day cholecalciferol ± 1 g omega-3 for 5.3 years, found a 22% reduction in confirmed incident autoimmune disease (HR 0.78, 95% CI 0.61–0.99) — the first randomized evidence that supplementation prevents rather than merely associates with lower autoimmune risk. Extended follow-up (Costenbader et al. Arthritis Rheumatol 2024;76:973–983) showed the effect attenuated after supplementation ended (HR 0.85, 95% CI 0.70–1.04), implying ongoing intake is needed.
Mechanistically, vitamin D converges on every pathway TCE disrupts: it upregulates Nrf2-driven antioxidant defenses (superoxide dismutase, glutathione peroxidase, catalase, glutathione synthesis), supports mitochondrial respiratory chain function, modulates VDR-expressing dopaminergic neurons, expands regulatory T cells while suppressing Th17/Th1 responses, and dampens dendritic cell maturation and B-cell autoantibody production. Glutathione depletion and GSH/GSSG redox imbalance are precisely the pathway TCE activates in the cerebellum in MRL+/+ mice (Blossom/Gilbert work). Other antioxidants tested against TCE neurotoxicity — N-acetylcysteine, vitamin E, coenzyme Q10 — show partial rescue in rodent models, but no published rodent or human study has specifically tested vitamin D supplementation against TCE-induced Parkinsonism or autoimmunity. A PubMed search for "vitamin D" AND "trichloroethylene" returns essentially no primary evaluation studies. The claim that vitamin D protects against TCE toxicity is therefore a mechanistically plausible but unproven hypothesis: reasonable to pursue in animal work, premature to offer as clinical advice.
What the evidence supports and where it breaks down
Reading across these literatures, TCE should now be regarded as one of the best-established environmental causes of Parkinson's disease, on par with paraquat and rotenone, and as a confirmed trigger of scleroderma, hypersensitivity syndrome and likely other autoimmune conditions, with strongest evidence in men occupationally exposed (for scleroderma) and HLA-B13:01+ Asian workers (for hypersensitivity). The quantitative signals — 1.7-fold PD risk at Camp Lejeune, 6-fold in twins with occupational exposure, 2-fold for scleroderma, 27-fold for HLA-B13:01 hypersensitivity — are large enough to drive public-health action even with exposure assessment imperfection. Key limitations remain: Camp Lejeune exposed people to multiple co-contaminants, making pure TCE attribution impossible; no quantitative dose-response meta-analysis exists for PD; animal doses are often higher than environmental exposures (though the 50-ppm inhalation Adamson 2023 data approach realistic levels); and the 2025 ambient-air finding rests on a single cross-sectional exposure proxy. Vitamin D's role against TCE is a research gap worth filling, not a conclusion. Finally, the December 2024 TSCA ban is necessary but not sufficient — ~850 Superfund sites, vast groundwater plumes, vapor intrusion into homes and the 50-year cleanup horizon mean that TCE exposure, and the Parkinson's and autoimmune cases attributable to it, will continue emerging through the 2050s and beyond. That latency is the reason the case for aggressive remediation, biomonitoring and presumptive compensation has only grown stronger since Goldman's 2023 paper reframed the problem.
Trichloroethylene and vitamin D
There's very little direct research on a TCE–vitamin D interaction, but there are several strong indirect links worth considering for VitaminDWiki.
Parkinson's disease — the clearest convergence
TCE is now considered an environmental driver of PD. A JAMA Neurology study of Camp Lejeune veterans showed 70% higher PD risk in those exposed to TCE-contaminated water compared with similar veterans stationed elsewhere, and occupational exposure has been linked to roughly a 500% increased PD risk in earlier work. A 2025 Medicare-based case-control study found a dose-dependent relationship between ambient TCE and PD incidence, with a relative risk of 1.10 comparing top to bottom decile of exposure.
Vitamin D sits on the protective side of the same disease. A meta-analysis of seven studies (5,690 PD patients, 21,251 controls) found deficient vitamin D more than doubled PD risk (OR 2.08), while supplementation reduced risk (OR 0.62) and outdoor work reduced risk (OR 0.72). Mechanistically, vitamin D appears to act on PD pathology through the VDR, CYP2D6, the renin-angiotensin system, heme oxygenase-1, PARP-1, nerve growth factor, L-type voltage-sensitive calcium channels, and antioxidant pathways.
Since TCE causes PD largely through cytochrome P450-generated reactive metabolites that disrupt mitochondrial function, induce oxidative stress, and activate neuroinflammation leading to dopaminergic neuron loss, vitamin D's anti-inflammatory and antioxidant actions plausibly counter the very mechanisms TCE uses. No RCT has tested this directly.
Autoimmune disease — a second convergence
TCE exposure has been associated with autoimmune effects in both mice and humans — increased antinuclear antibodies, increased IFN-γ, decreased IL-4, autoimmune hepatitis, and in three case-control studies a combined odds ratio of 2.5 for scleroderma in men occupationally exposed to TCE. Chronic TCE exposure has also been linked to systemic lupus, hepatitis, and diabetes, with CD4+ T cell alterations a common biomarker.
Vitamin D deficiency independently raises risk for most of those same conditions (lupus, scleroderma, autoimmune hepatitis, type 1 diabetes), so TCE and low vitamin D may act additively on a shared immunologic substrate.
Shared CYP metabolism — an underexplored angle
This is where a VitaminDWiki page could add real value. TCE is metabolized by two pathways: an oxidative pathway initiated by cytochrome P450s (primarily CYP2E1) and a glutathione conjugation pathway via GSTs. Vitamin D activation and catabolism also runs through the CYP system — CYP2R1 and CYP27A1 for 25-hydroxylation, CYP27B1 for activation, and CYP24A1/CYP3A4 for catabolism. Drugs that induce CYP3A4 accelerate vitamin D catabolism and can contribute to vitamin D deficiency. Whether chronic TCE exposure alters hepatic vitamin D hydroxylase activity (either by inducing competing CYPs or by liver damage from TCE metabolites) is not well-studied but is mechanistically plausible.
Glutathione connection
TCE's GSH conjugation pathway consumes glutathione and produces nephrotoxic metabolites (DCVG, DCVC). This ties into your prior glutathione work — populations with chronic TCE exposure likely have depleted glutathione reserves, and vitamin D is known to upregulate glutathione synthesis. This is another protective angle worth developing.
Bottom line for a VitaminDWiki page
The direct "does vitamin D level correlate with TCE toxicity outcomes" literature is essentially empty — I couldn't find a single study measuring 25(OH)D in TCE-exposed cohorts. But the indirect evidence (shared diseases, shared mechanisms, shared CYP machinery, oxidative/mitochondrial vulnerability that vitamin D mitigates) is substantial enough to justify a hypothesis-level page. Camp Lejeune veterans would be an obvious population to propose studying, given that many have ongoing VA care and 25(OH)D measurements in their records.
Trichloroethylene use in pulp plants (A friend of mine who worked in a pulp plant got PD
Yes, TCE has definitely been used in pulp and paper operations, though it wasn't the signature solvent of the industry (metal degreasing was its dominant use). The American Chemistry Council lists pulp and paper as one of the sectors where TCE has served as a solvent, and Australia's environmental agency specifically identifies pulp and paper manufacture — particularly de-inking of paper — as a primary source of TCE emissions. EPA guidance on the paper industry notes that wastewater and treatment sludges from mills can fail toxicity testing due to organic constituents including chloroform and trichloroethylene, which tells you it was present in the process stream. It would typically have been used for degreasing machinery, cleaning rollers and equipment, and de-inking in recycled paper operations — not in the pulping/bleaching chemistry itself.
The Parkinson's link is now about as well-established as environmental causation gets in this field. The landmark 2023 JAMA Neurology study of Camp Lejeune veterans found a 70% higher risk of Parkinson's in those exposed to TCE-contaminated water decades earlier, and an earlier epidemiological study had found occupational or hobby TCE exposure associated with a 500% increased risk. The cluster that kicked off much of the modern research was a Kentucky doctor in 2008 who realized three of his Parkinson's patients had all worked ~25 years at the same plant where they were exposed to TCE, with several coworkers showing early signs as well. Mechanistically, TCE inhibits mitochondrial complex 1 and destroys dopamine-generating neurons in the substantia nigra in exposed rodents — the same pathological signature seen with paraquat and rotenone. The VA has presumed a link between Parkinson's and occupational TCE exposure for Camp Lejeune service members since 2017, and the EPA announced a ban on nearly all TCE uses in late 2024.
Practically, this matters for your friend in two ways. First, if he's a U.S. worker, a pulp plant exposure history with a Parkinson's diagnosis is exactly the fact pattern that workers' comp and toxic tort attorneys are now pursuing — worth a consultation with an occupational disease attorney, and worth his neurologist documenting the occupational history in the chart. Second, there's growing interest in whether early/aggressive intervention on known PD modifiers (exercise, which he's already likely aware of given your VitaminDWiki work, plus the usual levodopa timing questions) may matter more in environmentally-caused PD. The TCE-PD literature is recent enough that a lot of treating neurologists haven't caught up with it — the Ray Dorsey paper "Trichloroethylene: An Invisible Cause of Parkinson's Disease?" is the single best thing to hand him or his doctor.
Global use of Trichloroethylene is decreasing (esp. if use more than 1000kg/year)
Quick note on the US status before getting to other countries: the December 2024 EPA rule technically bans all uses of TCE under TSCA, but the story is messier than "banned." The rule faced legal challenges and, after consolidation in the Third Circuit, the broad prohibitions took effect on March 28, 2025, except for provisions covered by critical use exemptions. EPA has repeatedly postponed the effective date for the Section 6(g) exemption conditions — most recently pushing them to May 18, 2026 — while the Third Circuit litigation continues. So some uses are prohibited now, some are operating under stayed conditions, and the final shape depends on the court.
For the rest of the world:
European Union (and EEA). The EU has effectively phased out most TCE use without a formal outright ban. The European Commission added TCE to the REACH Authorisation List (Annex XIV) in 2013 for all uses because of its carcinogenic effects, and from 2016 users were required to have an authorisation for continued use. Over 95% of uses within the scope of authorisation have been phased out in the EU since the substance was added to the Candidate List in 2010. The most common substitute in industrial metal parts cleaning has been perchloroethylene, which is itself toxic to aquatic life and suspected of causing cancer — a sideways move that researchers have criticized. A handful of specialized industrial applications still run under granted authorisations; new ones are rare.
United Kingdom. Post-Brexit, the UK operates UK REACH, which inherited the EU's authorisation requirements for TCE. The practical regulatory posture is similar: not a blanket ban, but authorisation required for continued use, with most industrial users already having switched.
Canada. Canada has not banned TCE outright but has regulated it tightly for over two decades. In 2003, Canada issued the Solvent Degreasing Regulations to reduce TCE releases from facilities using more than 1,000 kilograms per year, and in 2013, Canada added TCE to the Toxic Substances List — Schedule 1 under CEPA. TCE is not manufactured in Canada; domestic supply comes from imports, and the solvent degreasing regulations are designed to significantly reduce its use and release. The drinking water guideline is 5 ppb.
Japan. Under Japan's Chemical Substances Control Law, TCE is designated a Class II substance — one that may pose a risk of long-term toxicity to humans or to flora and fauna. That triggers manufacturing/import notifications and use controls rather than a ban. Japan also has occupational exposure limits and has seen steady substitution pressure.
Australia and New Zealand. TCE remains on national chemical inventories (AIIC in Australia) and is regulated as a hazardous workplace substance and dangerous good for transport, but there is no outright ban. Use has declined heavily through substitution and workplace exposure standards. New Zealand regulates it under the Hazardous Substances and New Organisms framework.
China and South Korea. Both maintain TCE on restricted or controlled substance lists with workplace exposure limits, reporting requirements, and some use restrictions. China is still a significant producer and consumer of TCE, particularly as a feedstock for HFC refrigerants — though that downstream demand is itself shrinking as HFCs are phased down under the Kigali Amendment.
Global context worth noting. TCE is not listed under the Stockholm Convention on POPs, so there is no binding international treaty banning it. The broad global pattern has been: IARC classified it as Group 1 (known human carcinogen) in 2012, and since then most developed economies have moved through a combination of workplace exposure limits, authorisation regimes, and substitution incentives rather than formal bans. The US 2024 rule is the most aggressive "outright ban" action by a major regulator to date — which is partly why it has drawn such heavy litigation.
One wrinkle relevant to continued global use: the main current use of TCE is as feedstock to produce other chemicals, such as fluorinated hydrocarbons and fluorinated polymers; about 80% of EU production goes to this use. Feedstock use in closed systems is typically treated more leniently than open solvent use across all these jurisdictions, so "banned" rarely means zero molecules moving through industry.
Liposomal Glutathione also fights TCE, unless you have a kidney problem
Yes — but the relationship is more nuanced than the usual functional-medicine narrative suggests, and the nuance matters for a VitaminDWiki page.
The simple story (mostly correct)
TCE metabolism consumes glutathione. The second major TCE metabolic pathway is conjugation with glutathione in the liver to form S-(1,2-dichlorovinyl)glutathione (DCVG), which is then converted to S-dichlorovinylcysteine (DCVC) primarily in the kidney. Chronic exposure therefore drains GSH reserves. In mice, postnatal TCE exposure via drinking water shifted hippocampal glutathione redox homeostasis toward a more oxidized state, increased the oxidative-stress biomarker 3-nitrotyrosine in plasma and hippocampus, altered transsulfuration and transmethylation pathway metabolites, and reduced expression of BDNF, NGF, and NT-3. In Drosophila, TCE significantly raised ROS and depleted total thiols while inhibiting catalase and glutathione S-transferase; co-administration of the flavonoid hesperidin restored these antioxidant markers.
On the clinical side, integrative practitioners do recommend liposomal glutathione specifically for TCE exposure support. Ann Shippy's protocol for TCE exposure describes Phase 1 support with milk thistle, vitamin C, sulforaphane, and cruciferous vegetables, and Phase 2 support with liposomal glutathione, noting that in Phase 2 the remaining TCE metabolites are conjugated with glutathione. There is no RCT testing liposomal glutathione against TCE-related outcomes in humans — this recommendation is mechanistic, not evidence-based.
The complication that matters
TCE's GSH pathway is a double-edged sword. The glutathione conjugate DCVG is itself the precursor to the nephrotoxic and nephrocarcinogenic metabolite DCVC. In a classic Lash study on isolated rat kidney and liver cells, increases in cellular GSH concentrations increased TCE-induced cytotoxicity in kidney cells but not hepatocytes, consistent with GSH conjugation driving nephrotoxicity; conversely, GSH depletion moderately decreased TCE toxicity in kidney cells but increased it in hepatocytes, pointing to different bioactivation pathways in the two organs.
So the likely picture:
- Liver, immune system, brain: GSH is protective. TCE depletes GSH, oxidative stress rises, damage follows. Repleting GSH (including liposomally) is plausibly beneficial.
- Kidney: more GSH means more DCVG, which means more DCVC, which means more tubule damage. This is the organ where TCE causes carcinogenesis through the GSH pathway.
This is an under-discussed tension. The functional-medicine literature almost universally recommends GSH for TCE "detox" without mentioning that the kidney — the primary target organ for TCE carcinogenesis — is harmed through glutathione conjugation, not in spite of it.
Liposomal specifically
Liposomal delivery is relevant because oral GSH is largely degraded in the gut. Liposomal glutathione may be more effective than standard oral glutathione, which is thought to be broken down by enzymes in the stomach. That's the bioavailability argument you've already covered on VitaminDWiki. The TCE question is whether better systemic delivery amplifies the good (hepatic/neural/immune protection) or the bad (renal DCVC formation) — and nobody has studied that directly.
The vitamin D tie-in for a VitaminDWiki page
Vitamin D upregulates glutathione synthesis (via Nrf2 pathway and GCLC expression), which links back to your earlier glutathione content. A TCE page could frame the argument as: chronic TCE exposure depletes systemic GSH → vitamin D sufficiency supports endogenous GSH regeneration → this likely protects liver, brain, and immune compartments but does nothing special for the kidney, which remains TCE's primary organ at risk. That's a more honest framing than the usual "take glutathione for detox" line, and it reinforces the case for vitamin D as the upstream, root-cause intervention rather than supplementing the end-product antioxidant.
One practical caveat worth flagging on any page you write: for people with known kidney impairment and ongoing TCE exposure, aggressive GSH repletion may not be the unambiguous good that the wellness industry implies.
Century-Old Cleaning Chemical Linked to 500% Increased Risk of Parkinson’s Disease
Related in VitaminDWiki
- Parkinson’s Disease and Vitamin D – review of 52 studies
- Parkinson's Disease 5X more likely if Trichloroethylene (TCE, wonder about Vit D)
- PM2.5 pollution associated with 9 types of deaths (all associated with low Vitamin D)
- Ovarian Cancer – increased risk near pulp mills)
- Brain health improved by Vitamin D - review of 90 studies
- Vitamin D and neurodegenerative diseases
- Glutathione fights: Diabetes, Alz, PD, NAFLD, Kidney, UC, IBD, HIV, Fatigue, etc,
- Glutathione fights Parkinson's Disease
- PFAS (forever chemicals) reduce Vitamin D and VDR, cause health problems - many studies
- 20+ chronic stressors that result in poor health
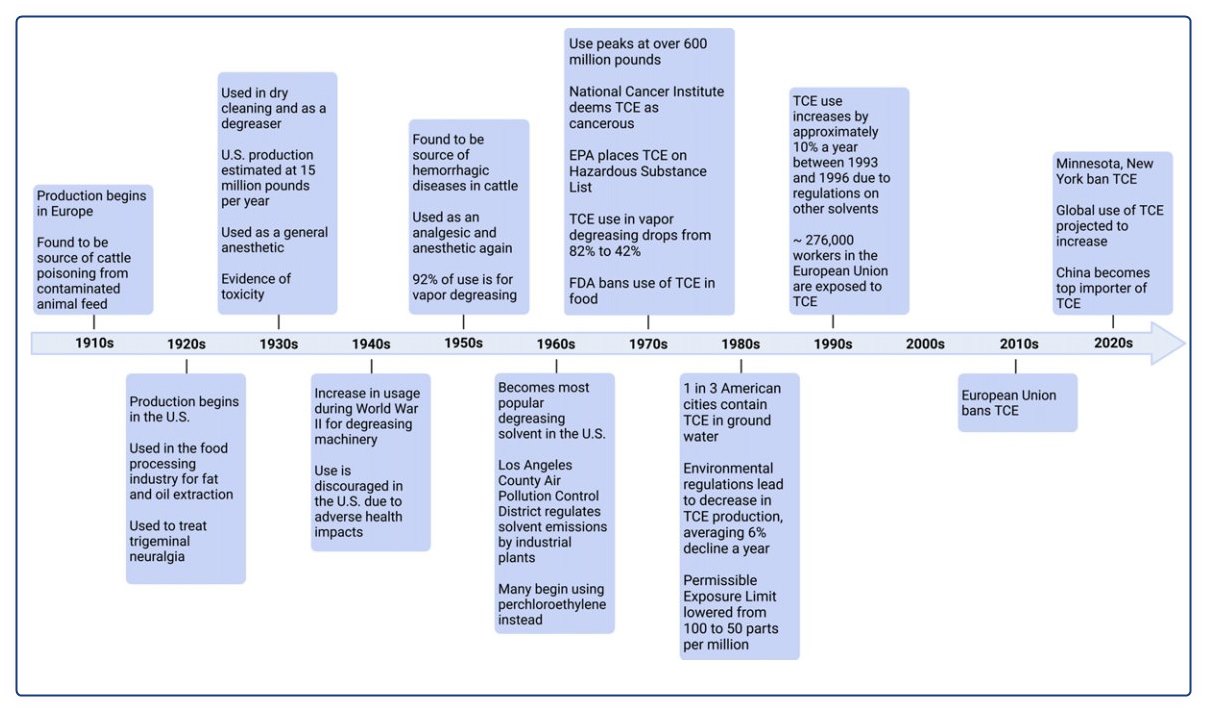
- Parkinson's Disease 5X more likely if Trichloroethylene contains the following timeline and lists
Adhesives, Aerosol cleaning products, Carpet cleaner, Cleaners and solvent degreasers, Cleaning wipes, Cosmetic glues, Decaffeinated coffee, Film cleaners, Glue, Gun cleaner, Fumigant, Hoof polishes, Inks, Lubricants, Mold release, Paint and paint removers, Pepper spray, Pesticides, Refrigerant, Sealants, Stain removers, Tap and die fluid, Toner aid, Tool cleaners, Typewriter correction fluids, Wood finishes,
Automotive care, Dry cleaning, Degreasing, Furniture care, Manufacturing, Computer and electronics, Disinfectants, Dyes, Fat and oil extraction, Flavor extracts (spices, hops), Jewelry, Machinery, Paint and coating, Paper, Perfumes, Plastics, Refrigerant*, Soaps,
Anesthesia (medical, dental, veterinary), Surgical disinfectant, Treatment (migraines, trigeminal neuralgia), Pharmaceutical manufacturing,
Aircraft maintenance workers, Automotive factory workers, Communications equipment repairers, Computer specialists, Corrosive control technicians, Distillery workers, Dry cleaners, Electronic component manufacturers, Embalmers, Food manufacturers, Insecticide manufacturers, Jet engine mechanics, Leather manufacturers, Machinery installation & assembly workers, Mechanics, Metal treatment workers, Missile technicians, Nautical equipment workers, Oil processors, Painters, Pesticide manufacturers, Pharmaceutical manufacturing factory workers, Printers, Radar technicians, Refrigerant manufacturers, Resin workers, Rubber cementers, Sewerage workers, Silk screeners, Shoe makers, Systems technicians, Taxidermists, Textile manufacturers, Textile and fabric cleaners, Tobacco denicotinizers, Waste treatment workers, Weapons specialists, Varnish workers