Fructose, HFCS and Vitamin D - many studies
Fructose, HFCS and Vitamin D: What the Evidence Actually Shows
Claude AI Deep Research June 2026
TL;DR
- The relationship is bidirectional but unevenly evidenced: rodent studies robustly show high fructose lowers the active hormone 1,25(OH)₂D₃ (calcitriol) by suppressing renal CYP27B1 and inducing CYP24A1 — but this has never been demonstrated in humans, and even in rodents the precursor 25(OH)D is usually unchanged.
- In the other direction, vitamin D co-treatment consistently protects fructose-fed rodents against fatty liver, insulin resistance and hypertension, yet human RCTs in NAFLD/metabolic syndrome are largely null for liver fat and inconsistent for metabolic endpoints.
- Human epidemiology linking sugar-sweetened beverages (SSB) to lower 25(OH)D is cross-sectional only, heavily confounded (BMI, diet quality, outdoor activity), and cannot establish causation; the mechanistic claims substantially outrun the clinical evidence.
Key Findings (tiered by evidence strength)
Tier 1 — Randomized controlled trials (humans): No RCT has ever tested whether fructose/HFCS intake changes vitamin D metabolites. RCTs of vitamin D supplementation in NAFLD/metabolic syndrome exist and are mostly null for hepatic fat; meta-analyses show modest improvements in insulin-resistance markers but not core liver outcomes.
Tier 2 — Prospective cohorts: None directly link fructose/SSB intake to change in 25(OH)D over time. This is a complete gap.
Tier 3 — Cross-sectional human: Several datasets (including NHANES 2013–2014) associate higher SSB/sugar intake with lower 25(OH)D, but effects are largely BMI-mediated and confounded.
Tier 4 — Animal/rodent mechanistic: Strong, reproducible work (Douard/Ferraris group) shows fructose lowers serum 1,25(OH)₂D₃, suppresses renal CYP27B1, induces CYP24A1, and impairs intestinal calcium absorption. Separate rodent studies show vitamin D protects against fructose-induced metabolic harm.
Tier 5 — In vitro/mechanistic: Supports plausibility (VDR effects on lipogenesis, gut tight junctions) but is hypothesis-generating only.
Details
Direction 1: Fructose/HFCS impairing vitamin D status
The foundational evidence comes from the laboratory of Veronique Douard and Ronaldo P. Ferraris at Rutgers/New Jersey Medical School, across several rodent papers.
Douard et al., J Am Soc Nephrol 2010;21:261–271 (PMC2834550): In 5/6-nephrectomized rats fed a 60% fructose vs glucose diet for 1 month, fructose reduced serum 25(OH)D₃ (by 30–40%) and 1,25(OH)₂D₃ (2.5–4-fold lower) — but only when renal function was already compromised. Fructose also markedly reduced intestinal calcium absorption, mediated mainly by decreased calbindin (CaBP-9k) expression. Notably, in this CKD model renal CYP27B1 (1α-hydroxylase) protein actually increased 2–5-fold while CYP24A1 was unchanged — the opposite enzymatic pattern to later studies, illustrating the model-dependence of these mechanisms.
Douard et al., Am J Physiol Endocrinol Metab 2013;304:E1303–E1313 (growing rats): Growing rats fed fructose (vs glucose or starch) for 4 weeks had reduced intestinal calcium transport, lower 1,25(OH)₂D₃, reduced bone length and bone ash. Dietary fructose increased CYP24A1 and decreased CYP27B1 expression, and serum FGF23 was upregulated, suggesting a bone-derived signal suppressing 1α-hydroxylase. Serum calcium, phosphate and PTH were unchanged. (An earlier paper, Douard et al., FASEB J 2012;26:707–721, showed the same in lactating rats with elevated calcium demand.)
Douard et al., PLoS One 2014;9(4):e93611 (PMC3981704) — the pivotal calcium-sufficient study: Adult mice/rats fed high fructose (43–63% of energy) with normal calcium showed reduced renal CYP27B1, increased CYP24A1, and reduced serum 1,25(OH)₂D₃ over 3 months. Critically, the precursor 25(OH)D₃ was NOT reduced — the effect was specific to the renal activation/degradation step. The authors state: "chronically high fructose intakes can decrease serum levels of 1,25(OH)2D3 in adult rodents experiencing no Ca2+ stress and fed sufficient levels of dietary Ca2+." Serum FGF23 rose over time but the increase was often not statistically significant ("FGF23 production did not increase…perhaps due to a statistically insufficient number of samples").
Doses are extreme. These rodent diets supplied 43–63% of energy as fructose — far above human intakes (~10% of calories). The authors explicitly flag this in their Limitations section, noting that "rodents and humans have different diets" and humans consume lower, mixed-carbohydrate fructose loads.
Mechanisms invoked: suppression of renal CYP27B1 (less calcitriol synthesis); induction of CYP24A1 (more catabolism); fructose-induced FGF23 elevation (which physiologically suppresses CYP27B1 and induces CYP24A1); reduced intestinal calbindin/calcium transporters. Effects on vitamin D binding protein specifically were not a focus and remain essentially unstudied in the fructose context.
Human evidence for Direction 1: none interventional. There is no controlled feeding trial or RCT measuring 25(OH)D or 1,25(OH)₂D before/after fructose, sucrose or HFCS, and no prospective cohort tracking change in 25(OH)D against fructose intake. Two subtle but important points: (1) the rodent effect is on calcitriol (1,25(OH)₂D₃) via CYP27B1, NOT on 25(OH)D — yet human cross-sectional studies measure only 25(OH)D, so they are not even testing the proposed mechanism; (2) acute human fructose ingestion raises FGF21 (a hepatic metabolic hormone; Dushay et al., Mol Metab 2015 reported a ~3.4-fold rise after 75 g oral fructose), NOT the phosphaturic FGF23 relevant to vitamin D. Fructose→FGF23 has not been tested in humans, and even the rodent FGF23 signal is inconsistent.
Direction 2: Vitamin D modulating fructose-driven metabolic harm
Rodent co-treatment studies (Tier 4) — consistently protective: - A Western (high-fat/fructose) diet study in rats (PMC6137584) found vitamin D₃ supplementation reduced steatotic hepatocytes (61% → 27%), roughly halved HOMA-IR (~41.9 → ~19.4), and lowered blood pressure and left ventricular mass — even without baseline vitamin D deficiency. - A high-fat + 25% fructose NAFLD rat model (Frontiers in Pharmacology 2023, PMC10228732) showed vitamin D₃ (1,000 IU/kg, 3×/week, 10 weeks) reduced hepatic SREBP-1c, restored PPAR-α, improved redox status and reduced steatosis/inflammation. - A fructose-specific study (Lipids in Health and Disease 2024) tested vitamin D₃ (10,000 IU/kg/week) ± high-intensity interval training against fructose-induced hepatic lipid accumulation, reducing de novo lipogenesis markers. - Calcitriol in fructose-fed hypertensive rats (PMC4361671; 60% fructose, calcitriol 10–20 ng/100 g/day) improved endothelial function, glucose tolerance and visceral adiposity. - HFCS brain study: Aslan et al., Behavioural Brain Research 2024;459:114763 — adolescent rats given an 11% HFCS-55 solution ± vitamin D (42 µg/kg/day) for 31 days; vitamin D reduced degenerative neurons in prefrontal cortex and improved learning/memory.
Mechanisms: suppression of hepatic lipogenesis (SREBP-1c), anti-inflammatory/antioxidant effects, improved gut-barrier integrity and reduced endotoxemia (calcitriol promotes intestinal tight junctions), and possible uric-acid effects.
Human evidence (Tier 1) — disappointing: - Barchetta et al., BMC Medicine 2016;14:92 (PMC4926287): RCT of cholecalciferol 2,000 IU/day vs placebo for 24 weeks in T2D patients with NAFLD; 25(OH)D rose significantly in the active arm (48.2 → 89.8 nmol/L) but there was NO change in MRI-measured hepatic fat fraction (between-group difference 0.3% [1.3%], p = 0.57), transaminases, CK18-M30, or metabolic/cardiovascular parameters. - Meta-analyses are mixed: one (PMID 32966467, 10 trials, 544 subjects) found vitamin D significantly reduced fasting glucose (−0.22), insulin (−0.68) and HOMA-IR (−1.32) but not AST, total cholesterol, HDL or LDL in NAFLD; others found null effects on core hepatic outcomes. - Vitamin D supplementation does NOT reliably improve lipid profiles; several RCTs (e.g., the Styrian Hypertension Study; Maki et al.) even reported triglyceride increases.
Crucially, none of these human trials used a fructose challenge — they test vitamin D in metabolic disease generally, not specifically fructose-driven disease.
Direction 3: Epidemiology and historical context
- HFCS consumption in the US rose more than 1,000% between 1970 and 1990 — Bray, Nielsen & Popkin, Am J Clin Nutr 2004;79:537–543: "The consumption of HFCS increased >1000% between 1970 and 1990, far exceeding the changes in intake of any other food or food group." Per-capita HFCS peaked at 65.9 lb/person/year in 1999 (USDA Economic Research Service) and has since steadily declined to 39.5 lb by 2021. (An older 2008 ERS "Amber Waves" availability series gives a 63.7 lb peak; the current ERS series gives 65.9 lb.)
- Population 25(OH)D appeared to fall between NHANES III and the 2000s — Ginde, Liu & Camargo, Arch Intern Med 2009;169(6):626–632: "The mean serum 25(OH)D level was 30 (95% CI, 29–30) ng/mL during NHANES III and decreased to 24 (23–25) ng/mL during NHANES 2001–2004." HOWEVER, this apparent decline is largely an assay artifact: Looker et al., Am J Clin Nutr 2008;88:1519–1527 (PMC2745830) found that most of the observed NHANES III → 2000–2004 difference "appears to be an artifact of assay changes rather than an actual decline," with assay adjustment accounting for much of the gap; only a small real decline persisted in a subgroup of non-Hispanic white adults (attributed to rising BMI, less milk, more sun protection). The "HFCS rise mirrors vitamin D decline" narrative is therefore largely unsupported once methodology is corrected.
- Cross-sectional associations: NHANES 2013–2014 (Zhou et al., Nutrients 2023;15:3291, PMC10421177; n=4,505) found heavy SSB consumers had ~2-fold higher odds of vitamin D deficiency (aOR = 2.10, 95% CI 1.25–3.54), with ~21.3% of the effect mediated by BMI. A cross-sectional premenopausal-women study (Nutrients 2014, PMC4145290; mean plasma 25(OH)D 65.0 nmol/L, 23.2% below 50 nmol/L) found higher cola intake associated with lower 25(OH)D. These are correlational and confounded.
What the Evidence Does NOT Show (research gaps)
- No human proof that fructose lowers vitamin D. Zero interventional or prospective human data. The mechanism is rodent-only at supraphysiological doses.
- Mechanism/measurement mismatch. Rodent data implicate calcitriol/CYP27B1; human studies measure only 25(OH)D, which rodents show is largely unaffected by fructose.
- Reverse causation and confounding unresolved. Heavy soda drinkers tend to be heavier, less active, spend less time outdoors, and eat worse overall — all independently lowering 25(OH)D. BMI is a major mediator/confounder.
- The ecological correlation is weak. Once assay drift is corrected, US vitamin D status did not actually fall during the HFCS surge.
- Human protective trials are null for the headline outcome (liver fat), and vitamin D may worsen triglycerides.
- FGF23/Klotho pathway untested in humans with fructose; rodent FGF23 findings are inconsistent, and human acute-fructose studies measure FGF21, a different hormone.
- Vitamin D binding protein effects of fructose are essentially unstudied.
Recommendations
For readers/clinicians: - Reducing fructose/SSB intake is justified on well-established grounds (obesity, NAFLD/MASLD, dental, metabolic) independent of any vitamin D claim. - Do not expect vitamin D supplementation to reverse sugar-driven fatty liver based on current human evidence. - Maintaining adequate 25(OH)D remains sensible general advice but should not be marketed as an antidote to a high-sugar diet.
Benchmarks that would change these conclusions: - A controlled human feeding study showing fructose lowers 1,25(OH)₂D₃ or raises CYP24A1 activity/catabolism. - A prospective cohort linking baseline fructose intake to subsequent 25(OH)D decline after adjusting for BMI and lifestyle. - An adequately powered RCT of vitamin D in biopsy-confirmed, fructose-driven NAFLD/MASLD with histological endpoints.
Caveats
- Much of the supporting literature is rodent work at fructose doses (43–63% of energy) that humans never reach; extrapolation is speculative and acknowledged as such by the original investigators.
- Some material circulating on this topic comes from secondary sources (reviews, news outlets, advocacy pages); the primary findings here are anchored to the Douard/Ferraris rodent papers and named human RCTs/NHANES analyses.
- The vitamin D/NAFLD RCT literature is heterogeneous in dose, duration and population; "null" reflects the best-quality trials (e.g., Barchetta 2016) but not unanimity — some smaller trials and meta-analyses report metabolic (not hepatic) benefit.
- Vitamin D itself is not uniformly benign in metabolic contexts: some rodent and human data show high-dose vitamin D can raise blood pressure or triglycerides, so "more is better" is not supported.
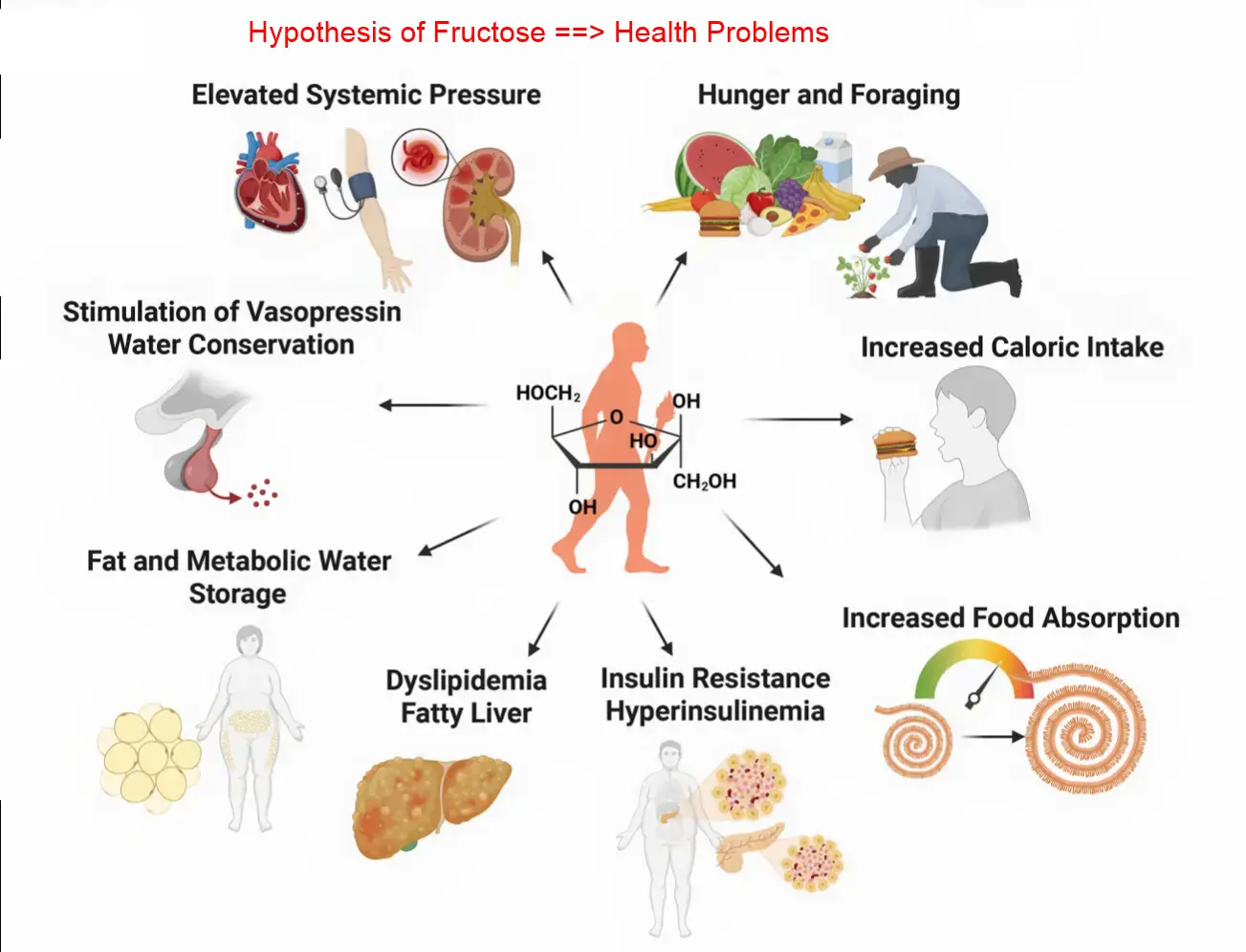
Fructose: Metabolic signal and modern hazard - May 2026
Vitamin D supplement reduced some HFCS problems in rats - Feb 2024
Protective effect of vitamin D on learning and memory impairment in rats induced by high fructose corn syrup
Behavioural Brain Research Volume 459, 29 Feb 2024, 114763
https://doi.org/10.1016/j.bbr.2023.114763 PDF is behind a paywall
Cahide Aslan a 1, Rahime Aslankoc a, Ozlem Ozmen b, Buse Nur Sülük a, Oguzhan Kavrık a, Nurhan Gumral a
In our study, we aimed to investigate the negative effects of the prefrontal cortex (PFC)-associated impairment of cholinergic activity on memory and learning caused by high fructose corn syrup (HFCS) and the protective role of vitamin D in adolescent rats.
Twenty-four animals were divided into three groups as control, HFCS group (11 % HFCS-55 solution, ad libitum) and HFCS+ Vit D (42 μg/kg/day). Elevated Plus Maze (EPM), Forced Swim Test (FST), and Morris Water Maze (MWM, performed from day 23) tests were applied to all animals. Fluid intake consumption of the rats was measured daily, weight gain and blood glucose were measured weekly. After 31 days of treatment, the rats were sacrificed and PFC tissue was removed for biochemical, histopathological and immunohistochemical analyses.
In HFCS group, fluid consumption, blood glucose, malondialdehyde (MDA) levels, degenerative neuron count and choline acetyltransferase (ChAT) expression were significantly increased; superoxide dismutase (SOD), catalase (CAT) enzyme activity and brain-derived neurotrophic factor (BDNF) expression were significantly decreased. In addition, the time spent in the enclosed arm in EPM was increased, the immobility time in FST was, and the time spent in the target quadrant in MWM was significantly decreased. Vitamin D treatment reversed all these parameters. In conclusion, HFCS caused an increase in the number of degenerative neurons in the PFC, disrupted cholinergic activity and negatively affected learning-memory functions. Vitamin D, decreased the number of degenerative neurons, increased cholinergic activity and positively affected learning and memory performance.
Brief Synopsis
In this study, prefrontal cortex damage was investigated in adolescent rats fed high fructose corn syrup. The effect of vitamin D on prefrontal cortex damage was evaluated.
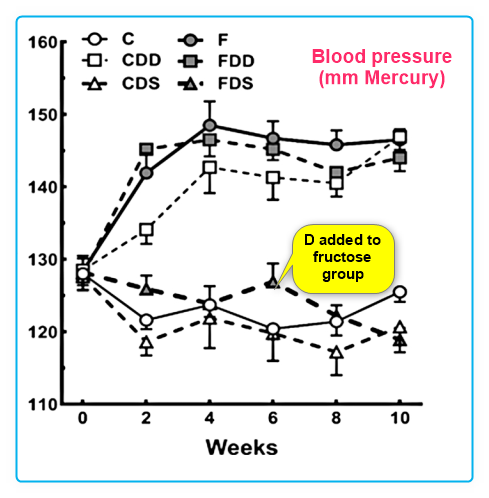
Vitamin D supplementation reduced problems of fructose-rich diets in mice - Sept 2019
The deficiency and the supplementation of vitamin D and liver: Lessons of chronic fructose-rich diet in mice
J of Steroid Biochemistry & Molecular Biology, Vol 192, Sep 2019, https://doi.org/10.1016/j.jsbmb.2019.105399
Thais C. Maia-Ceciliano, Rafaela R. Dutra, Marcia B. Aguila, Carlos A. Mandarim-De-Lacerda 1
Highlights
Vitamin D deficiency is growing at a global level and is linked to chronic diseases.
Fructose intake has a role in the epidemics of obesity and metabolic syndrome.
Fructose consumption plus vitamin D deficiency lead to steatosis and liver fibrosis.
Vitamin D supplementation lessen the adverse effects of the fructose intake.
The fructose added to soft drinks and processed food, as well as frequent detection of vitamin D deficiency in the body, are two insults increasingly considered to cause lesions in target organs. We studied the liver after a chronic high-fructose diet deficient and supplemented with vitamin D. Sixty C57BL/6 mature male mice were allocated into six groups (n = 10) for ten weeks: control (C), control deficient in vitamin D (CDD), control supplemented with vitamin D (CDS), fructose (F), fructose deficient in vitamin D (FDD), and fructose supplemented with vitamin D (FDS). The gene expressions of vitamin D receptor and CYP27B1 and 25 hydroxyvitamin D plasma level ensured that the diets caused vitamin D deficiency or supplementation. Body mass did not change, but blood pressure (BP) increased in CDD, F, and FDD, whereas BP was controlled in FDS. Insulinemia, insulin tolerance and resistance were seen in both vitamin D deficiency and fructose groups but improved with vitamin D supplementation. The steatosis and fibrosis were observed in the CDD, F and FDD groups. Also, F and FDD showed activation of stellate cells (HSC). Lipogenesis and inflammation gene expressions were enhanced in the CDD, F and FDD groups, but diminished with vitamin D supplementation. In conclusion, we demonstrated the adverse effects of vitamin D deficiency on metabolism, liver steatosis and, combined with fructose intake, liver interstitial fibrosis with hepatic stellate cell activation, and alteration of the lipogenesis, beta-oxidation, and liver inflammation. All these data improved when vitamin D was supplemented in the animals. 📄 Download the PDF from VitaminDWiki
Scientists claim to have found root cause of obesity for most people (HFCS) - Oct 2023
Daily Mail reporting on The fructose survival hypothesis as a mechanism for unifying the various obesity hypotheses
https://doi.org/10.1002/oby.23920 79 references online $12 to rent the PDF
The experts explained when people ingest fructose-heavy foods the amount of usable energy available to support the body’s cells plummets, leading to feelings of hunger.
Most carbs and fats people eat replace levels of ATP, a molecule that fuels cells so they can move, divide, and perform basic functions in the human body needed for survival.
This prompts the release of a hormone called leptin, which signals to the brain it’s time to stop eating.
But when fructose is metabolized in the liver, it uses ATP as an energy source. This causes fuel levels to plummet while at the same time interfering with the body’s ability to use stored fat as energy.
The reduction in ATP in cells is associated with hunger, thirst, increased food intake, reduced metabolism at rest, increased salt absorption, and more, all of which can lead to weight gain.
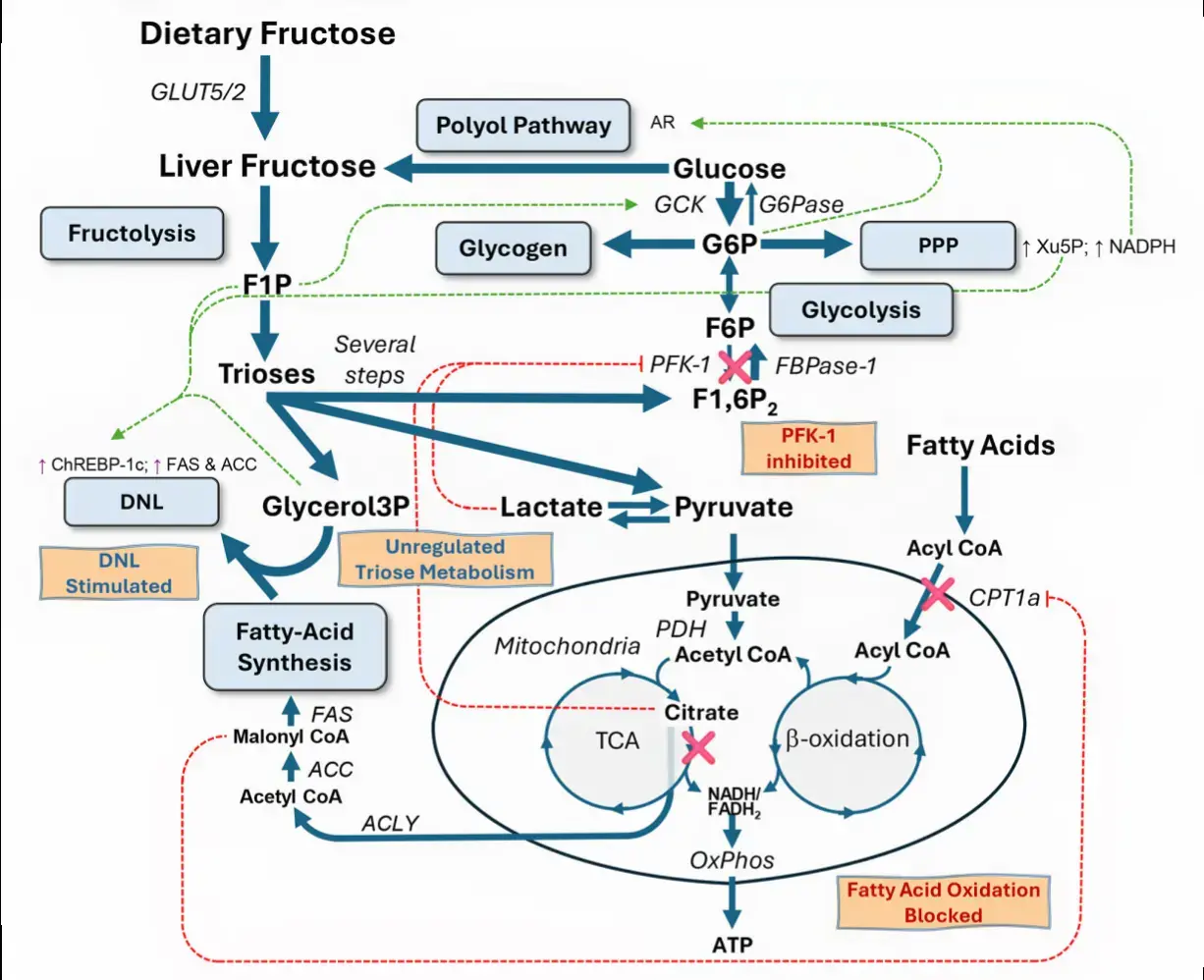
Fructose drives de novo lipogenesis affecting metabolic health - March 2023
in Journal of Endocrinology DOI: https://doi.org/10.1530/JOE-22-0270
Bettina Geidl-Flueck, Philipp A Gerber
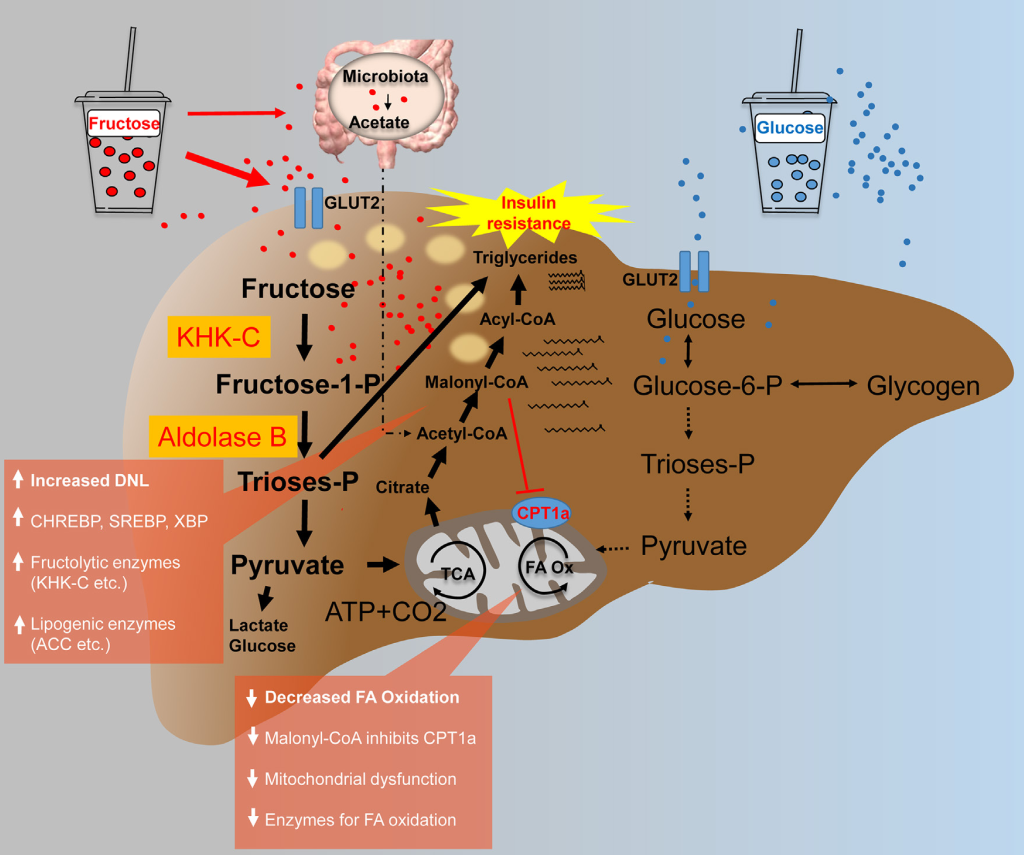
Despite the existence of numerous studies supporting a pathological link between fructose consumption and the development of the metabolic syndrome and its sequelae, such as non-alcoholic fatty liver disease (NAFLD), this link remains a contentious issue. With this article, we shed a light on the impact of sugar/fructose intake on hepatic de novo lipogenesis (DNL), an outcome parameter known to be dysregulated in subjects with type 2 diabetes and/or NAFLD. In this review, we present findings from human intervention studies using physiological doses of sugar as well as mechanistic animal studies.
There is evidence from both human and animal studies that fructose is a more potent inducer of hepatic lipogenesis than glucose.
This is most likely due to the liver’s prominent physiological role in fructose metabolism, which may be disrupted under pathological conditions by increased hepatic expression of fructolytic and lipogenic enzymes.
Increased DNL may not only contribute to ectopic fat deposition (i.e. in the liver), but it may also impair several metabolic processes through DNL-related fatty acids (e.g. beta-cell function, insulin secretion, or insulin sensitivity).