Parkinson's Disease perhaps prevented and treated by Vitamin D
Parkinson's Disease and the "Sunshine" Vitamin
Alzheimers Dis Parkinsonism 2013, 3:2 http://dx.doi.org/10.4172/2161-0460.1000120
Lucy Elisabeth James1 and Ayodeji A Asuni2 a.a.asuni@soton.ac.uk
1 Biomedical Sciences Undergraduate Program, Centre for Biological Sciences, Institute for Life Sciences, University of Southampton, Southampton, United Kingdom
2 Centre for Biological Sciences, Institute for Life Sciences, University of Southampton, Southampton, United Kingdom
Accrued evidence suggesting that hypovitaminosis D acts as a risk factor for developing Parkinson's disease (PD) remains controversial. Herein we evaluated existing results, and outline several biological mechanisms by which the hypovitaminosis D-PD relationship may occur. We performed a meta-analysis, using data obtained from a search of PubMed from July 2002 to July 2012, for studies reporting serum vitamin D levels in PD and control patients.We found that in comparison to healthy individuals, those with PD had lower levels of serum vitamin D. Furthermore, we explore a number of potential associated biological mechanisms, including the actions of reactive nitrogen species (RNS), glutathione (GSH), and glial-derived neurotrophic factor (GDNF) in the brain. We also examine the roles of Nurrl and VDR genes in PD. Although a unifying hypothesis remains challenging, there is evidence to demonstrate that supplementation with the vitamin can to have a positive effect on PD pathobiology. We surmise that hypovitaminosis D does act as a risk factor in the development of PD. However, the need for new epidemiological studies and further research around vitamin D metabolism is highlighted. Urgent efforts to correct vitamin D deficiency through supplementation are warranted as they may improve either motor and/or non-motor symptoms in PD.
PDF is attached at the bottom of this page
See also VitaminDWiki
- Overview Parkinson's and Vitamin D contains the following lsummary
{include}
Introduction
Globally, it is estimated that 7.4 million people have Parkinson's disease (PD). In the UK alone, a diagnosis occurs every hour, and among the elderly population, Parkinson's is the second most common type of neurodegenerative disease [1]. There are several forms of PD; however, in over 90% of cases, the etiology is unknown. For the remaining 10% of Parkinson's patients, it is recognised that they have monogenic forms of the disease. In familial Parkinson's, alterations may occur in genes such as SNCA or DJ-1 [2]. Identification of rare genes such as these is likely to give insight into the molecular pathways that underlie the disease. Most individuals who develop PD are 60 years of age or older; however 5% of cases are reported before the age of 50. The primary symptoms of PD are caused by dopamine insufficiency due to progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), leading to reduced stimulation of the motor cortex by the basal ganglia [3,4]. The depletion of dopamine is most prominent in the dorsolateral putamen, which is the main site of projection for these neurons [5]. By the time of death, up to 70% of neurons within the SNpc may be affected [6]. The decline in dopamine, causes symptoms such as tremor, rigidity, and bradykinesia, which are collectively termed 'parkinsonism' [7]. As the disease progresses, cognitive and behavioural problems may also arise. A second feature of PD pathology is the abundance of intraneuronal cytoplasmic inclusions in the brain, termed 'Lewy bodies'; which contain aggregates of various proteins and lipids and accumulate in the spared dopaminergic neurons of SNpc.
Microglia, the brains resident macrophages play a critical role in the maintenance of CNS microenvironment. One attractive hypothesis is that PD is partly driven by the inflammatory response of microglia [8,9], with activated microglia culpable for majority of tissue destruction in the PD brain. A strong inflammatory response is elicited in the cells upon encounter with extracellular stimuli, such as those from pathogens or dying neurons. Once activated, microglia educe a reaction in order to mediate the innate immune response and eliminate proinflammatory signals. Not only do these cells generate such mediators, but they also act to propagate the pro-inflammatory response, which activates further microglia and astrocytes. Direct stimulation of microglial populations with LPS and nitrated a-synuclein, have been observed to lead to the destruction of dopaminergic SNpc neurons both in vivo and in vitro [10]. While others have demonstrated increased levels of reactive oxygen species (ROS), reactive nitrogen species (RNS) and pro-inflammatory cytokines associated with by activated glial cells [11].
Another candidate implicated in PD pathogenesis is the ubiquitin-proteasome system (UPS) [7]. This system plays an important role in removing proteins that are no longer required inside cells. Failure of this system, from genetic and/or environmental factors, leads to the abnormal aggregation of proteins such as a- synuclein, the major component of Lewy bodies. Formation of Lewy bodies may lead to the activation of pathways, which later result in neuronal death.
Presently treatment options for PD are underwhelming. While it is possible to treat the symptoms of PD, it is not possible to reverse the effects of the disease or cure it. There are three main pharmacological drugs given for PD; Levodopa, dopamine agonists, and MAO-B inhibitors. The actual approach chosen varies depending on the stage of the disease. When a patient presents with PD symptoms for the first time, Levodopa (a precursor of dopamine) may be given. However, it is preferable for other medications such as MAO-B inhibitors and dopamine agonists to be administered prior to Levodopa in the hope that this will delay the onset of dyskinesia. If a patient develops motor complications that are related to Levodopa intake, then the physician's aim is to reduce symptoms of PD whilst controlling fluctuations of the response to medication. If medication is no longer able to control PD symptoms, then surgical techniques such as deep brain stimulation may be relied upon. Other established risk factors for PD are advanced age, a positive family history and environmental influences, which are controversial. There is some evidence that exposure to low dose organochlorine (OC) pesticides may lead to vitamin D deficiency in humans [12]. This warrants that chemical exposure as a possible cause of vitamin D deficiency should be evaluated in prospective and experimental studies.Lately hypovitaminosis D has been proposed as a significant risk factor in the development of PD [13, 14]. While the biological mechanisms behind the associations remain to be clarified, several theories have been put forward. Herein we outline the biology of vitamin D and prevalence of hypovitaminosis D as it relates to PD and discuss evidence that supports this.
Biology of the "Sunshine" Vitamin
The two main physiologically relevant forms of vitamin D are vitamin D2 and D3 (Figure 1). In humans, the latter is photosynthesised in the skin from 7-dehydrocholesterol (provitamin D3) following exposure to UVB light with a wavelength of 290 to 315 nm. The initial product (previtamin D3) is subsequently isomerised in a temperature dependent reaction to generate a more stable form of the vitamin [15]. Alternatively, vitamin D3 may be obtained from the diet, and is present in oil-rich foods such as herring, mackerel, and salmon. Vitamin D2, in contrast, is found naturally in sun-exposed fungi. The vitamin is synthesized fromviosterol, which becomes activated when UV light stimulatesergosterol. For the purpose of this review "vitamin D" may represent D2 and/or D3.
Vitamin D derived from the skin or diet is biologically inert regardless of its source and therefore undergoes hydroxylation during a two-step enzymatic pathway to produce the active form of the vitamin, 1,25-dihydroxyvitamin D (1,25(OH)2D) [16].Vitamin is metabolised in the liver (by vitamin D 25-hydroxylase, orCYP2R1) to produce 25-hydroxyvitamin D (25(OH)D). 25(OH)D is slowly released into the blood from the liver to achieve a concentration that is related to the total liver storage. 25(OH)D is then converted by 25-hydroxyvitamin D 1-a- hydroxylase (CYP27B1) in the kidneys to generate 1,25(OH)2D (Figure 2).
Figure 1:
Chemical structures of vitamin D2 and D3 (A) Structure of vitamin D2 otherwise known as ergocalciferol. (B) Structure of vitamin D3, which may also be referred to as cholecalciferol. Chemically, the two forms of the vitamin are related in that they are both secosteroids. Secosteroids are steroids in which one of the double bonds in the steroid ring is broken. Structurally, vitamin D2 and vitamin D, differ due to their side chains. The side chain of D, contains a double bond between carbon positions 22 and 23, and a methyl group on carbon 24.
Figure 2:
Vitamin D metabolism pathway. Ingested vitamin D, be it either vitamin D2 or D3 is incorporated into lipoprotein particles known as chylomicrons. These are transported by the lymphatic system into venous circulation, where the vitamin binds to the vitamin D binding protein (DBP). Skin derived vitamin D is also carried in the circulation by DBP. Vitamin D is transported to the liver where it is converted by vitamin D-25-hydroxylase to 25-hydroxyvitamin D (25(OH)D). In this form, the vitamin is biologically inactive and is therefore converted in the kidneys by 25-hydroxyvitamin D-1a-hydroxylase to ',25-dihydroxyvitamin D (',25(OH)2D). The chemical structures of 25(OH)D and l,25(OH)2D are shown in the figure. In the diagram, 'vitamin D' represents either vitamin D2 or D3.
Serum calcium and phosphorous levels, and plasma parathyroid hormone concentrations tightly regulate the renal production of 1,25(OH)2D. Once more, 1,25(OH) 2D may decrease its own synthesis through negative feedback. Prior evidence suggested the brain's supply of 1,25(OH)2D depended upon the plasma concentration of the active vitamin. However, recent evidence suggests otherwise; it is now understood that the CNS is capable of generating the active form of the vitamin. This is supported by the fact that both vitamin D25-hydroxylase and 25-hydroxyvitamin D-1a-hydroxylase are located within the brain. In vitro microglial are able to produce the active form of vitamin D from its precursor, and when exposed to 1,25(OH)2D, these cells can up-regulate the vitamin D24-hydroxylase gene. This gene encodes an enzyme that produces the inactive form of vitamin D. These discoveries therefore suggest that the brain has both synthetic and degradative pathways for the vitamin, allowing homeostasis [17].
In the presence of the active form of vitamin D, renal and intestinal calcium are absorbed far more efficiently; in the absence of 1,25(OH)2D, only ~10% of dietary calcium is taken up [18]. The regulation of calcium channels is an example of the fast actions of vitamin D, which occur in seconds. In addition, 1,25(OH)2D may exhibit its actions through a slower process that modulates the transcription of a number of genes (Figure 3). The biological actions of 1,25(OH)2D range from the inhibition of cellular proliferation, to the induction of insulin production. In fact, the local production of 1,25(OH)2D may be responsible for the regulation of around 200 genes[16].An individual's vitamin D status may be determined by the concentration of 25(OH) D in the blood rather than the level of 1,25(OH)2D; there are multiple
Figure 3:
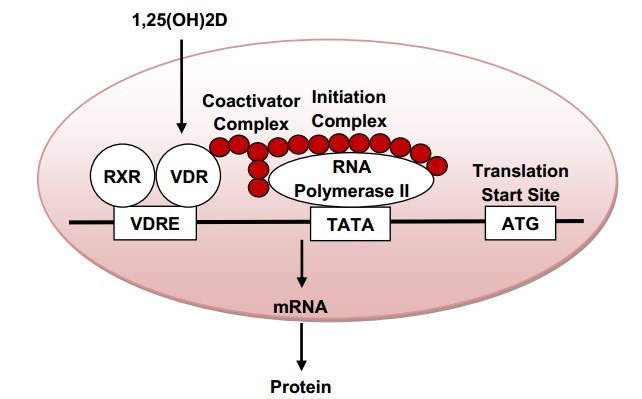
Regulation of vitamin D target genes. Circulating ',25(OH)2D passes into a target cell and binds to the cytoplasmic vitamin D receptor (VDR), which is present in most cells and tissues in the body. This translocates to the nucleus and in turn, interacts with nuclear hormone receptors, most principally, the nuclear retinoic acid X receptor (RXR). The ',25(OH)2D-VDR-RXR complex then binds to vitamin D response elements (VDREs) on DNA. This binding is accompanied by the formation of large complexes that are able to either facilitate the expression of a target gene (coactivators) or alternatively, inhibit its expression (cosuppressors). Cosupressors act predominantly by deacetylating histones. Coactivators, in contrast, function either to expose the gene for transcription, or to bridge the gap between the VDREs and initiation complex, which stimulates transcription by activating RNA polymerase. The horizontal black line in the figure represents DNA.
reasons for this, for example, 25(OH)D has a greater circulating half-life[19]. Some report qualify concentrations below 25 nmol/L (equivalent to 10 ng/ml) as 'deficient' vitamin D levels [20],while others contend that serum 25 (OH)D concentrations below 80 nmol/L (32 ng/ ml) are inadequate [21]. There is currently no standard definition of the 'optimal' 25(OH)D level, but suggested concentrations range from above 50 nmol/L (20 ng/ml) to 100 nmol/L (40 ng/ml)[20, 22]. Despite this, the UK Department of Health has set dietary recommendations for pregnant (or lactating) women, children below the age of 4 years old, and individuals over the age of 64 years old (or confined to the indoors); the Current Reference Nutrient Intakes are 10 |ig (400 IU) of vitamin D per day, 7-8.5 |ig (280-340 IU) per day, and 10 |ig (400 IU) of vitamin D per day, respectively [23].
Prevalence of Hypovitaminosis D
An estimation ofvitamin D deficiency as 50 nmol/L (20 ng/ml) serum 25(OH)D or below and vitamin D insufficiency as 52-72 nmol/L (20.828.8 ng/ml) serum 25(OH)D, would mean that an estimated 1 billion people worldwide suffer from vitamin D deficiency or insufficiency [18]. Suggesting the scale of the problem is enormous. Among the numerous factors, influencing cutaneous production of vitamin D3 is a geographic location north or south of the equator, the degree of skin pigmentation, or the lack of sun exposure due to religious coverings. Predominantly the major cause of vitamin D deficiency is due to inadequate exposure to sunlight. In addition, vitamin D deficiency that is not related to a lack of sun exposure is often observed in newborns, obese individuals, and those with a poor diet, individuals with liver or kidney disease and those with fat malabsorption [16,24]. It is evident that the amount of UVB in sunlight changes significantly depending on the season, latitude, and time of day. In a recent study, Hypponen et al. demonstrated that during the summer and autumn, 3.2%, 15.4%, and 60.9% of adults in Britain had 25(OH)D levels below 25, 40, and 75 nmol/L respectively. During winter and spring however, these figures increased to 15.5%, 46.6%, and 87.1% correspondingly [25]. Over the winter months, the UK population does not receive UV light of a suitable wavelength to synthesise vitamin D; for individuals living at latitudes of approximately 52° and above (Britain is between latitudes of 50° to 60° north) they must rely solely on body stores and dietary vitamin D. However, the advent of winter is not the only problems; as during summer months; application of sunscreen with a sun protection factor of 30 (SPF30) decreases photosynthesis of vitamin D by over 95% [16]. Doctors state that when the UV index is greater than 3, sun exposure (without sunscreen) of ten minutes, twice a week, will allow for an adequate amount of vitamin D to be synthesised.
Individuals with naturally dark skin are also at risk of vitamin D deficiency. To synthesize the same amount of vitamin D as someone with a light skin tone, those with darker skin tones need to expose themselves to UVB rays for three times longer; due to the increased amount of melanin in darker skin [26]. People who are homebound due to their age or disability, and women who cover their skin for religious purposes, are further examples of groups who are at an increased risk of deficiency due to inadequate sun exposure. Elderly people are also likely to suffer from deficiency, as individuals of an older generation are unable to synthesize vitamin D as efficiently as a younger generation; this is partially due to the age-related decline in skin thickness [27]. Thus, adequate sun exposure is very important in order to prevent vitamin D deficiency. Despite this, multiple studies have demonstrated that 25(OH)D concentrations plateau at around 70-80nmol/L after 'good UV exposure. In a Hawaiian study for example (latitude 21°), it was found that for a group of young individuals with no health problems, the mean serum 25(OH)D concentration was just 31.6 |g/L (79 nmol/L), despite excessive sun exposure [28]. Vitamin D deficiency causes abnormalities in calcium, phosphorous, and bone metabolism. In early life, deficiency may result in growth retardation and skeletal deformities, and in later life, it can cause osteomalacia and increase the risk of fracture. Recently, hypovitaminosis has also been heavily implicated in diseases such as cancer, heart disease, depression, as well as in MS [16,19,29-31].
PD and the "Sunshine" Vitamin: The Relationship
Meta-analysis
Numerous studies have been carried out to highlight the possible relationship between hypovitaminosis D and PD. In this report, we performed a meta-analysis to evaluate these data. The analysis was based on a search of PubMed from July 2002 to July 2012, using the key terms 'Parkinson's disease and vitamin D' or 'Parkinson's disease and vit D'; this initially yielded 92 papers. The inclusion criteria for the meta-analysis was as follows; (1) papers must compare one PD group to one control group; (2) in every paper the PD and control category require 50 or more subjects each; (3) papers must be written in English; and (4) lastly the studies must display 25(OH)D mean and standard deviation values for both PD and control groups, in addition to the information shown in Table 1. Based on these strict criteria 5 studies from the original 92 were included in the analysis, with a total of 434 Parkinson's disease cases and 3451 healthy controls[13,29,32-34]. The information taken from each paper and the results from the statistical tests performed are presented in Table 1 and Table 2, respectively.
While there is no definitive value for vitamin D deficiency or insufficiency, vitamin D deficiency can be assumed to be a value ofbelow 50 nmol/L (20 ng/ml) serum 25(OH)D , and vitamin D insufficiency 52-72 nmol/L (20.8-28.8 ng/ml) serum 25(OH)D [18]. Based on these values four of the five PD groups demonstrate a mean deficient vitamin D level. In addition, two of the five control studies display a mean deficient level, one an insufficient level, and two sufficient levels.
Conversely, if it is accepted that vitamin D deficiency is a lower value than the one proposed above however, for example below 37.6 nmol/L (15 ng/ml), then yet again four of the five PD groups still demonstrate a mean deficient value, whereas none of the control groups qualify as deficient in vitamin D by these criteria. Our analysis reveal that that patients with Parkinson's disease had decreased levels of 25(OH)D in comparison to the healthy controls (95% CI: -2.44 to -0.21; summary SMD: -1.33) (Figure 4).
The possible heterogeneity in the results was also examined; if the P value was greater than 0.05 and the I -squared value was less than 50%, the study was not considered to report significant differences. Results for this meta-analysis provided a p<0.001and an I -squared value of 98.2% which therefore displays a statistically significant heterogeneity. There is evidence to reject the null hypothesis that the true effects are identical. The significantly different heterogeneity may be due to the different ethnicities of the individuals in the study; the various countries that the individuals are from; or due to the varying amount of sunlight that each individual received. Overall we contend that subjects with Parkinson's disease had lower levels of 25(OH)D than the healthy controls.
Figure 4:
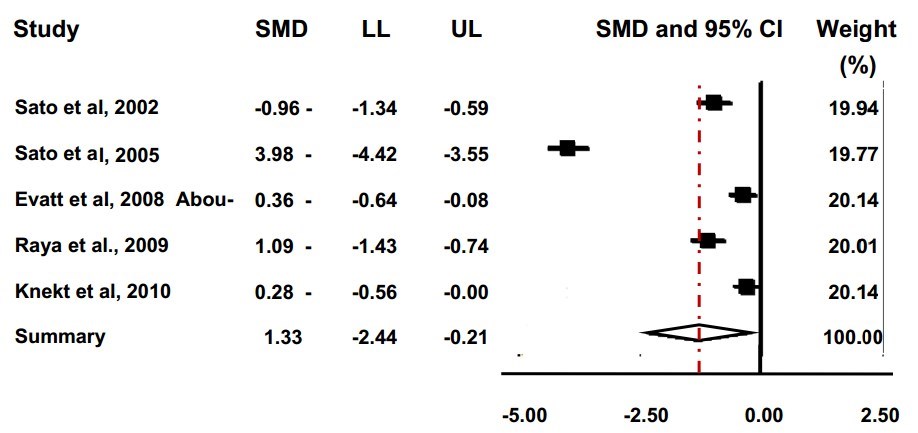
Forest plot to display meta-analysis results. The forest plot demonstrates the standardised mean difference (SMD), the confidence intervals (CI), and the weighting of each study. The SMD is represented by the squares and the 95% CI is shown as the whiskers on the graph. The diamond at the bottom of the plot symbolises the pooled estimate based on the random effects model. The centre of the diamond is used to demonstrate the point estimate, and the width of the diamond represents the associated 95% CI. LL and UL represent the lower and upper limit respectively.
PD and Vitamin D: Biological Evidence for a Relationship
Evidence for a biological relationship continues to accumulate, with some studies reporting that vitamin D exerts its effects on the disease through genomic and non-genomic mechanisms [1]. Due to the size of the relevant literature, several examples have been selected for discussion. The sections below point to the actions of reactive nitrogen species (RNS) and glutathione (GSH) reduction in the CNS; the VDR and glial-derived neurotrophic factor (GDNF); and lastly the Nurr1 gene.
Vitamin D, Inducible Nitric Oxide Synthase, Peroxynitrite and Glutathione
Reactive nitrogen species are a family of antimicrobial molecules; the initial reaction in which these are formed occurs between nitric oxide (NO) and the superoxide anion (O2-). NO is a highly reactive signalling molecule and is produced from L-arginine in a reaction catalysed by nitric oxide synthases (NOSs). In mammals, NO production is mediated by the calcium/calmodulin controlled isoenzymes endothelial nitric oxide synthase (eNOS) and neuronal nitric oxide synthase (nNOS). In addition, when stimulated by appropriate factors such as LPS and IFN-y for example, an inducible isoform (iNOS) produces NO and related RNS. Post mortem analysis of PD patients revealed an increased expression of iNOS in midbrain glial cells, with morphology characteristics of activated macrophages[35]. Interestingly, synthesis of iNOS has been shown to be inhibited by vitamin D; in experimental allergic encephalomyelitis models, 1,25(OH)2D down-regulates the expression of iNOS in the rat CNS [36, 37]. This raises the question of whether vitamin D deficiency could result in the up-regulation of iNOS expression, leading to an increased production of NO and RNS (and thus damage); this would provide a potential biological link between hypovitaminosis D and PD.
Table 1.
Data extracted from five studies comparing the 25(OH)D serum level (ng/ml) between individuals with Parkinson's disease and healthy control individuals.
Table 2.
Results from statistical tests that were performed for a meta-analysis comparing 25(OH)D serum levels (ng/ml) between individuals with Parkinson's disease, and healthy control individuals.
The RNS peroxynitrite (ONOO-) is generated from the reaction between NO and -O2 and its formation is regulated by the NO concentration in the body. It is a potent oxidant and nitrating agent that can react with other molecules to generate additional RNS such as nitrogen dioxide (-NOJ and dinitrogen trioxide (NO) or alternatively may interact with biological targets, for instance neighbouring neurons and astrocytes. Peroxynitrite is capable of attacking and altering proteins, lipids, and DNA, in addition to depleting the body's antioxidant defences. The anion may modify proteins by interacting with a variety of amino acids; for example, it is able to nitrate tyrosine residues to create 3-nitro-L-tyrosine. This resulting nitrophenol is harmful via a number of mechanisms; firstly, because nitrotyrosine bears a resemblance to phosphotyrosine, it can irreversibly block tyrosine phosphorylation, and therefore affect tyrosine phosphorylation dependent signalling. Presumably, autoimmune processes cannot be ruled out, as nitrophenols, such as 3-nitro-L-tyrosine, are exceptionally antigenic. Lastly, the alteration of protein conformation and augmentation of proteolysis through the introduction of a negative charge in hydrophobic tyrosine may occur. ONOO- has also been observed to induce lipid peroxidation, and cause DNA strand scission [38].
In addition, glutathione reductase, an enzyme that regenerates glutathione from its oxidised form (GSSG), is susceptible to peroxynitrite. A peroxynitrite concentration of 0.09 mM is sufficient to inhibit glutathione reductase by half [39]. The antioxidantglutathione (GSH), plays a critical role by preventing damage to cellular components that would otherwise result from the actions of reactive oxygen and nitrogen species.
Figure 5:
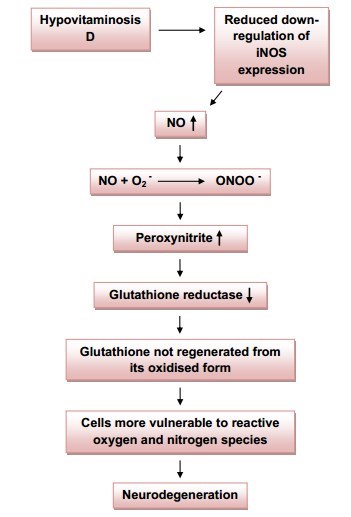
Potential biological mechanism behind the hypovitaminosis D-PD relationship.',25(OH)2D) can down-regulate the expression of inducible nitric oxide synthase (iNOS). Therefore it follows that hypovitaminosis D may lead to more nitric oxide (NO) being produced due to an increase in iNOS. NO is able to react with superoxide anion (O2-) to form peroxynitrite (ONOO-), which is capable of causing harm to cells (not shown), generating additional RNS (not shown), and inhibiting the activity of glutathione reductase, an enzyme that regenerates glutathione from its oxidised form (GSSG). Thus, less of the anti-oxidant glutathione will be present with an increase of peroxynitrite. Cell damage and neurodegeneration may then proceed; this suggests a vicious cycle in which the death of neurons leads to microglial activation, which in turn potentiates neuronal damage.
Seeing as peroxynitrite leads to the depletion of the antioxidant glutathione, this suggests that the RNS may render dopaminergic neuronal cells (that are already susceptible to a high oxidative load) more vulnerable to toxic insult, as glutathione would no longer be available in sufficient quantities to counter its actions (Figure 5). A decrease in intracellular glutathione from the substantia nigra is observed even in very early stages of PD [39]. Interestingly, vitamin D has also been said to be involved in the expression of the antioxidant, and is able to enhance the concentration in the CNS, presumably by stimulating some of the steps of glutathione synthesis [40, 41]. Thus, vitamin D appears to control brain detoxification processes and when sufficient, is able to significantly reduce toxic damage. This idea is supported by studies performed on rat cultured mesencephalic dopaminergic neurons treated with the inhibitor of glutathione synthesis L-buthionine sulfoximine (BSO) [42]. Hypovitaminosis D may thus directly and indirectly result in the depletion of glutathione in the CNS, leading to neurodegeneration.
Vitamin D, the Vitamin D Receptor, and Glial-Derived Neurotrophic Factor
The VDR is widely expressed in the human brain, and is present in both neurons and glial cells [43]. Within the brain, the highest expression is in hypothalamus and large neurons (likely to be the dopaminergic neurons) of the SN [44]. VDR is a member of the nuclear receptor family of transcription factors. As mentioned previously, when activated by vitamin D, VDRs interact with nuclear hormone receptors, and bind to VDREs on DNA. This leads to the regulation of expression of gene products. Studies that have sought to establish the role of the VDR in neuronal cells have demonstrated that vitamin D is involved in the induction of GDNF both in vitro and in vivo [45]. GDNF has protective effects against a range of toxic insults and promotes the survival of dopaminergic neurons. For example, it is up regulated by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrine (MPTP). MPTP is selectively toxic to cells in the SN; as such, MPTP-induced Parkinsonism animal's models can reproduce some clinical features of idiopathic PD. Moreover, the administration of GDNF reduces MPTP toxicity. Thus, the induction of GDNF expression during neuronal damage suggests that this protein may act as a neuroprotective agent. It is interesting to speculate whether hypovitaminosis D downregulates GDNF expression, leading to loss of its neuroprotective effect on dopaminergic neurons. On a side note, the VDR gene itself has been implicated in PD in addition to the effects mediated through the receptor. Butler et al. for instance, performed a two-stage study to evaluate the genetic effects of VDR in PD. The group examined VDR polymorphisms in a large Caucasian cohort of PD families, and confirmed the connection using genome-wide association study (GWAS) data. The paper highlights that the VDR gene may cause susceptibility to Parkinson's, however the exact mechanisms relating VDR single nucleotide polymorphisms (SNPs) and Parkinson's disease are not yet clear. It is concluded that common genetic variations in VDR modulate age-at onset of PD [2].
Vitamin D and the Nurrl Gene
The Nurr1 gene is another possible link between vitamin D deficiency and PD. Nurr1 is a member of the nuclear receptor family of intracellular transcription factors and the protein is essential for the development, survival, and functional maintenance of dopaminergic neurons within the midbrain [1]. For this reason, the Nurr1 gene was suggested to be involved in the pathogenesis of PD.Le and colleagues measured the relative expression of Nurrl in blood peripheral lymphocytes (PBL) for individuals with PD, and for healthy control patients, and reported that it was significantly decreased in the brain of PD patients [46]. Reduced levels of the gene were associated with a substantially increased risk of PD in patients who were 60 years or older, female, and of Caucasian origin [46]. A second study confirmed that decreased Nurr1 expression was linked to increased risk for PD in Chinese males over the age of 50 [47]. These data suggests that Nurr1 may be systemically involved in PD pathogenesis and possibly progression of the disease. It is possible that vitamin D deficiency could lead to decreased Nurrl expression, and in turn impaired survival of the dopaminergic neurons; as presented in the PD brain.
PD and Vitamin D: Evidence Against a Relationship
Very little evidence exists that challenges the hypovitaminosis D-PD relationship. It is interesting to note however, that many studies claim PD is more common in people with white skin colour, and less common in people with black skin colour. If the hypothesis relationship between hypovitaminosis D and PD were correct, one would most likely expect the opposite to be true, considering that people with black skin colour are more prone to vitamin D deficiency. Whether or not PD frequency varies by race has been a source of controversy for many decades. Mayeux et al. reported that in a population in Manhattan, the greatest incidence of PD was among people with black skin colour [48], but such are of the minority and many others contradict them [49]. Willis and colleagues performed a serial cross-sectional study of US Medicare beneficiaries aged 65 or older. The group analysed data on more than 450,000 cases of PD per year over six years (1995 and 2000-2005), and age, race, sex, and county calculated PD prevalence and annual incidence. It was established that both the incidence and prevalence of PD is higher in people with white skin colour, compared to those with black skin colour [50].
In addition, even among individuals of the same race, a lighter skin colour was found to be related to a higher risk of PD. In a case control study in California that involved 509 newly diagnosed PD patients, it was demonstrated that darker skin colour was inversely associated with PD risk among Caucasians and African-Americans [51].
These findings are important to consider when evaluating the strength of the hypovitaminosis D-PD relationship theory. Burne et al. behaviourally characterised VDR knockout mice (KO) by subjecting them to a battery of tasks. The KO mice were observed to be less active in the open field and buried fewer marbles in the marble burying test. Despite this, the mice did not demonstrate a difference in cognition; they exhibited no impairments in exploration, working memory, or anxiety. The group therefore claimed that VDR KO mice display a number of PD symptoms such as motor and muscular impairments, yet exhibit normal cognitive function [52]. This is of note, as cognitive function declines for many PD patients as the disease progresses.
Discussion
Meta-analysis
Results from the meta-analysis clearly demonstrate that subjects with PD have lower levels of 25(OH)D in comparison to healthy controls. Which we take as validation of the hypothesis that hypovitaminosis D is contributory to PD pathogenesis. Strengths of the meta-analysis were that it uses a large sample size of 434 Parkinson's disease cases and 3451 healthy controls, and each input paper had 50 or more subjects in both the PD group and control group; this gives confidence to the conclusions drawn. Furthermore, there were no anomalies, and each of the five studies reached the same conclusion. There are though limitations to the meta-analysis, which we acknowledge. Firstly, despite the large sample size, the meta-analysis used data from only four countries, and therefore it is not possible to assume the findings stand for the whole world. Data from other countries may still contradict our observations. In addition, there was some difference in the method used measure the vitamin D serum levels in some of the studies; a weakness also acknowledged by the studies themselves. For example Evatt et al. state that the portion of plasma samples drawn in winter-spring and autumn-summer were not matched across cohorts [13]. Whilst Knekt et al. remarked that it was possible that their study population might include some undefined PD cases [34]. Our analysis included a cross sectional study, such as Sato et al. as well as a longitudinal study, like Knekt et al. The appeal of these two types of studies varies. A longitudinal study tends to involve a repeated observation of the same variables over a long period. Due to this, a great amount of in-depth data is able to be accumulated. However, studies such as these are very time consuming and are expensive to perform. In contrast, a cross-sectional study usually observes a representative population at only one specific point in time. Despite this, it is still possible to compare two or more independent cross-sectional studies, from different times and samples, if available.
Conscious that our original meta-analysis search yielded few studies, we performed a second search of Pubmed using the same inclusion criteria as before, but from 1971 (earliest date available) to July 2002. No additional studies were found that could have been included in the meta-analysis based on our strict criteria. However, within this period there were studies that compare one control group to more than one PD group; but they demonstrated a lower level of serum 25(OH)D in the control group in contrast to the PD groups[53,54]. Remarkably, for such an important issue, extremely little robust data exists even when the search extends to in excess of 40 years prior.
Vitamin D and PD: Biological Evidence for and Against
Although, evidence of role for hypovitaminosis D in PD suggests there are number of potential mechanisms in which deficiency could contribute to the pathogenesis of PD as we outlined earlier. Further studies are required to fully address these claims, and we propose some experimental studies below 'Future Perspectives'.While we are satisfied that the meta-analysis and weight of biological mechanisms demonstrates there is a relationship between hypovitaminosis D and the risk of PD, we are also not overlooking what little evidence there is against the relationship. We are unable to rule out the possibility of 'file drawer bias', whereby our hypothesis is disproven, but the data simply remained unreported. One such caveat is the unexpectedly evidence, was the study reporting that the prevalence and incidence of PD was higher for those with white skin colour in comparison to those with black skin colour. This is an important finding, considering that people with black skin colour are more likely to be deficient in vitamin D due to increased levels of melanin in their skin. Whether the susceptibility of hypovitaminosis D varies between persons of lighter or darker skin remains the subject of much research. Interestingly, it was recently demonstrated that lower serum 25(OH)D levels are associated with an increased risk of incident coronary heart disease in white and Chinese subjects, yet a similar relationship is not observed among Black or Hispanic individuals. The estimates of 25(OH)D in ethnically homogeneous populations may not be broadly applicable to other racial or ethnic groups [55]. Regardless, when addressing the hypovitaminosis D-PD relationship theory, it is important to note that many other factors are responsible for causing vitamin D deficiency, especially at an older age. For this reason, the reporting that PD is more common in people with white skin does not negate any of our conclusions. Interestingly Burne et al. demonstrated that VDR KO mice exhibit normal cognitive function, while others disagreed [56,57]. Lead us to query whether vitamin D was more involved in the motor aspects of PD, and less in cognitive dysfunction. The exact cause of cognitive decline in PD patients is unknown, but Lewy bodies and deficits in neurotransmitter systems are thought to play a causative role; while dysfunction of dopaminergic, cholinergic, noradrenergic and serotonergic systems have been implicated. Further studies with VDR KO mice may provide a clearer answer to these questions.
Clinical Trials
Supplementation with vitamin D appears to have a beneficial effect on PD [1]. In a very recent study, the result of vitamin D supplementation was observed for patients with the VDR polymorphism FokI [58]. In a previous paper, the same authors stated that increased 25(OH)D levels and the VDR FokI CC genotype may be independently associated with milder forms of PD. The present study included 114 patients with Parkinson's and was performed over one year. Of these subjects, half were given 30 |ig (1200 IU) of vitamin D3 per day, and half were given a placebo each day. The authors found that compared to the placebo, supplementation significantly prevented deterioration of PD for this short period of time. This conclusion was only drawn for patients with FokITT (homozygous "variant") and FokI CT (heterozygous) genotypes however, and not for the CC genotype [58].
To the best of our knowledge, this is the first clinical trial to determine the effects of vitamin D supplementation on PD that also included a control group. Despite this, a recent meta-analysis demonstrated that supplementation with 20-25 |ig (800-1000 IU) vitamin D per day resulted in positive effects on strength and balance [59]. Thus, although Suzuki et al. suggest that supplementation may have beneficial effects on PD (for certain VDR polymorphism genotypes), one is not able to distinguish from this study whether supplementation specifically delays PD progression, or whether it simply improves muscle strength and balance. Supplementation with vitamin D has also been trialled for its effect on poor cognitive function, another symptom of PD. Dhesi et al. reported that when patients with vitamin D insufficiency were randomised to receive a single intramuscular injection of 15,000 |g (600,000 IU) vitamin D2 (rather than a placebo), this had a significant beneficial effect on functional performance, reaction time and balance after six months. The group suggest that vitamin D supplementation may therefore improve neuromuscular or neuroprotective function [60]. From studies of this nature, it appears that vitamin D deficiency may be detrimental to executive functions, however other cognitive domains such memory may be relatively preserved [61].
Preceding these trials, Derex and Trouillas recorded the details of a patient with PD who was admitted to hospital at the age of 50 years. The patient had a serum 25(OH)D concentration of 33 nmol/L (13.2 ng/ml) and no underlying condition that could cause this deficiency. Thus, the patient was given 100 g (4000 IU) of vitamin D3 and 1 g of calcium supplements daily in addition to ongoing anti-parkinsonian therapy (Levodopa 750 mg daily, bromocriptine, 20 mg daily, and trihexyphenidyle 10 mg daily). This resulted in the patient's 25(OH) D levels normalising. Once more, the Parkinsonism improved significantly during the following year. At a one year follow-up, the anti-parkinsonism drugs had been restricted to 375 mg of Levodopa daily, and neurological examination revealed moderate rigidity without tremor [62].
Hence, this study appears to follow the hypovitaminosis D-PD relationship theory. However, seeing as the clinical trial included only one person, this does not give an accurate representation of whether the same results would be obtained for the whole population. Once more, the patient was originally deficient in calcium, and therefore the calcium supplements may have acted in part, or completely, to improve the PD symptoms. Calcium participates in many interactions in the brain and particularly in the SNpc (the region that is heavily affected in PD).Thus, large scale clinical trials to confirm that vitamin D supplementation are beneficial for individuals with PD are very much needed. A trial is currently underway at Emory University and is due to be completed in December 2013 with an estimated enrolment of 150 individuals. Since vitamin D is recognised as an essential vitamin, the ongoing VIDIP pilot treatment study (http://clinicaltrials.gov/ct2/ show/NCT00571285) bypass potential ethical issues associated with restricting PD patents from vitamin D by designing their study such that subjects were assigned to either high dose vitamin D supplement (54,200 IU weekly) or the Recommended Daily Allowance (RDA) for older persons (4200 IU weekly of vitamin D) plus a placebo capsule. Subjects are to be examined in the clinic before, and then 3 and 6 months after taking the supplements. There is hope that this trial will provide evidence that is much needed.
Of the trials mentioned above, some used a vitamin D2 supplement whereas others used a vitamin D3 supplement. Equimolar quantities of vitamins D2 and D3 are assumed to be biologically equivalent, but Heaney et al. recently demonstrated that vitamin D3 is approximately 87% more potent in raising and maintaining 25(OH)D concentrations[63]. Therefore, supplementation with vitamin D3 may be more effective in trials compared to D2.
Conclusions
Four key points that support the hypovitaminosis D-PD relationship hypothesis. Firstly, the highest expression of the VDR in the brain is found within the substantia nigra, the region that is mainly affected in PD. Secondly, animal and human studies have demonstrated that vitamin D is an important agent for normal brain function. Hypovitaminosis D may lead to neurodegeneration in the SN, and cause greater sensitivity to toxic species such as RNS. Third, our meta-analysis shows a connection between the development of PD and vitamin D deficiency; every one of the five studies documented lower 25(OH)D levels in individuals with PD compared to control subjects. Lastly, to support this, clinical trials suggest that vitamin D supplementation has a beneficial effect on both cognition and motor symptoms of PD. However, large-scale clinical trials are needed as very few currently exist.
The precise role of hypovitaminosis D in PD pathogenesis remains unresolved, but the biological plausibility of this relationship is acknowledged and support by research evidence. Below we highlight three areas of research that require more attention; epidemiological studies, experiments relating to the biological mechanisms proposed above, and clinical trials.
Epidemiological Studies Present
There is an urgent need for more epidemiological studies to be carried out. Our study compared evidence from just four populations, and although the sample size was large, this could be increased significantly, rendering conclusions more accurate. It would be interesting to perform longitudinal studies in a number of countries spread across the equator, using the same methods of data accumulation and analysis (so that studies could be compared) to determine whether deficient 25(OH)D levels contribute to the development of PD. Details such as the ethnic background of the subjects within each country must be documented for example, to allow for those of the same origin to be grouped. This would mean that data from each study could be compared easily, and trends would become more obvious. However, geopolitical instability in several countries presents one potential obstacle for globe initiation of these studies.
Proposed Novel Experimental Paradigms
Vitamin D up-regulates glutathione, but it would be interesting to determine the differential effect of high and low concentrations of vitamin D on GSH in in vivo models. It also would be interesting to establish whether the lack of GSH, due to the actions of peroxynitrite on glutathione reductase, could be attenuated by de novo synthesis of GSH, or whether such mechanisms would also be impaired. In addition, vitamin D has the ability to inhibit the expression of iNOS. Although it has been proposed that this may reflect the presence of a VDRE in the promoter of the iNOS gene, this is not completely understood and consequently studies are needed to determine this. Moreover, it would be interesting to examine whether GDNF could be up-regulated in animal models of neurodegenerative disease if the rodent was deficient in vitamin D. Finally, to address the controversy that currently exists; new studies are required to establish the effect that VDR KO has in rodents. Although typically considered a disease of the brain, PD also affects a number of bodily systems including the enteric system. Future studies should therefore also aim to understand the impact of low vitamin D status on the digestive system [64].
Clinical Trials
Additional clinical trials are required to observe subjects from different backgrounds; examining a single population is not sufficient. Moderate supplementation for those with sufficient serum 25(OH) D levels would also be informative, as to date the focus has been on patients that are vitamin D deficient. It would be worthwhile to perform a study to determine whether there is a certain age category for which supplementation appears to have the greatest effect at slowing the development of PD (if at all). Perhaps there is a stage of Parkinson's which is too advanced for supplementation to have a positive effect. Furthermore, for each clinical trial, if large enough cohorts were acquired, it may be advisable to use vitamin D2 supplements for a third of the subjects, vitamin D3 for the second third and a placebo for the final third. This would allow one to establish whether vitamin D3 has a greater effect on PD than vitamin D2.
Most importantly, appropriate supplementation dosing studies are needed. Trials with too smaller dose could give a false negative result and trials with a dose of vitamin D that is too great could be toxic to the participants; interestingly, in experiments with rat cultured mesencephalic neurons, high doses of vitamin D were toxic to cells rather than beneficial. Holick states that intoxication is observed when 25(OH) D serum levels are above 374 nmol/L (150 ng/ml)[18]. High doses of vitamin D could also result in a hypercalcemic effect; it is important that researchers are aware of this before undertaking clinical trials. Lastly, trials should be performed over a relatively long period of time, as short trials will not determine whether supplementation is completely safe being as minor clinical signs and symptoms could be missed.
Using already published data, we examined the hypothesis that hypovitaminosis D contributes to PD disease pathogenesis, and evaluated potential biological mechanisms by which this may occur. Based on research evidence this hypothesis holds true, and efforts to correct hypovitaminosis D through vitamin D supplementation warrant attention for PD and potentially other chronic neurodegenerative diseases.